Jay S. McDonald-Ramos , Ian K. Hicklin , Zhaomin Yang , Anne M. Brown
{"title":"通过集合虚拟筛选鉴定嗜热绿脓杆菌 IV 型 Pilus 蛋白 PilB 的小分子抑制剂。","authors":"Jay S. McDonald-Ramos , Ian K. Hicklin , Zhaomin Yang , Anne M. Brown","doi":"10.1016/j.abb.2024.110127","DOIUrl":null,"url":null,"abstract":"<div><p>Antivirulence strategy has been explored as an alternative to traditional antibiotic development. The bacterial type IV pilus is a virulence factor involved in host invasion and colonization in many antibiotic resistant pathogens. The PilB ATPase hydrolyzes ATP to drive the assembly of the pilus filament from pilin subunits. We evaluated <em>Chloracidobacterium thermophilum</em> PilB (<em>Ct</em>PilB) as a model for structure-based virtual screening by molecular docking and molecular dynamics (MD) simulations. A hexameric structure of <em>Ct</em>PilB was generated through homology modeling based on an existing crystal structure of a PilB from <em>Geobacter metallireducens</em>. Four representative structures were obtained from molecular dynamics simulations to examine the conformational plasticity of PilB and improve docking analyses by ensemble docking. Structural analyses after 1 μs of simulation revealed conformational changes in individual PilB subunits are dependent on ligand presence. Further, ensemble virtual screening of a library of 4234 compounds retrieved from the ZINC15 database identified five promising PilB inhibitors. Molecular docking and binding analyses using the four representative structures from MD simulations revealed that top-ranked compounds interact with multiple Walker A residues, one Asp-box residue, and one arginine finger, indicating these are key residues in inhibitor binding within the ATP binding pocket. The use of multiple conformations in molecular screening can provide greater insight into compound flexibility within receptor sites and better inform future drug development for therapeutics targeting the type IV pilus assembly ATPase.</p></div>","PeriodicalId":8174,"journal":{"name":"Archives of biochemistry and biophysics","volume":"760 ","pages":"Article 110127"},"PeriodicalIF":3.0000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S0003986124002492/pdfft?md5=8129562d1f8578e6c794166cd6c848c7&pid=1-s2.0-S0003986124002492-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Identification of small molecule inhibitors of the Chloracidobacterium thermophilum type IV pilus protein PilB by ensemble virtual screening\",\"authors\":\"Jay S. McDonald-Ramos , Ian K. Hicklin , Zhaomin Yang , Anne M. Brown\",\"doi\":\"10.1016/j.abb.2024.110127\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>Antivirulence strategy has been explored as an alternative to traditional antibiotic development. The bacterial type IV pilus is a virulence factor involved in host invasion and colonization in many antibiotic resistant pathogens. The PilB ATPase hydrolyzes ATP to drive the assembly of the pilus filament from pilin subunits. We evaluated <em>Chloracidobacterium thermophilum</em> PilB (<em>Ct</em>PilB) as a model for structure-based virtual screening by molecular docking and molecular dynamics (MD) simulations. A hexameric structure of <em>Ct</em>PilB was generated through homology modeling based on an existing crystal structure of a PilB from <em>Geobacter metallireducens</em>. Four representative structures were obtained from molecular dynamics simulations to examine the conformational plasticity of PilB and improve docking analyses by ensemble docking. Structural analyses after 1 μs of simulation revealed conformational changes in individual PilB subunits are dependent on ligand presence. Further, ensemble virtual screening of a library of 4234 compounds retrieved from the ZINC15 database identified five promising PilB inhibitors. Molecular docking and binding analyses using the four representative structures from MD simulations revealed that top-ranked compounds interact with multiple Walker A residues, one Asp-box residue, and one arginine finger, indicating these are key residues in inhibitor binding within the ATP binding pocket. The use of multiple conformations in molecular screening can provide greater insight into compound flexibility within receptor sites and better inform future drug development for therapeutics targeting the type IV pilus assembly ATPase.</p></div>\",\"PeriodicalId\":8174,\"journal\":{\"name\":\"Archives of biochemistry and biophysics\",\"volume\":\"760 \",\"pages\":\"Article 110127\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S0003986124002492/pdfft?md5=8129562d1f8578e6c794166cd6c848c7&pid=1-s2.0-S0003986124002492-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Archives of biochemistry and biophysics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0003986124002492\",\"RegionNum\":3,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/16 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Archives of biochemistry and biophysics","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0003986124002492","RegionNum":3,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/16 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
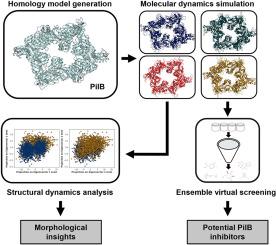
人们一直在探索抗病毒策略,以替代传统的抗生素开发。细菌 IV 型柔毛是一种毒力因子,参与了许多抗生素耐药病原体对宿主的入侵和定植。PilB ATP 酶水解 ATP,驱动柔毛亚基组装柔毛丝。我们通过分子对接和分子动力学(MD)模拟评估了嗜热绿杆菌 PilB(CtPilB),将其作为基于结构的虚拟筛选模型。根据现有的来自 Geobacter metallireducens 的 PilB 晶体结构,通过同源建模生成了 CtPilB 的六聚体结构。通过分子动力学模拟获得了四个具有代表性的结构,以研究 PilB 的构象可塑性,并通过集合对接改进对接分析。模拟 1 μs 后的结构分析表明,单个 PilB 亚基的构象变化取决于配体的存在。此外,对从 ZINC15 数据库中检索到的 4,234 种化合物进行了集合虚拟筛选,确定了五种有前景的 PilB 抑制剂。利用 MD 模拟的四个代表性结构进行的分子对接和结合分析表明,排名靠前的化合物与多个 Walker A 残基、一个 Asp-box 残基和一个精氨酸指相互作用,表明这些残基是 ATP 结合口袋内抑制剂结合的关键残基。在分子筛选中使用多种构象可以更深入地了解化合物在受体位点内的灵活性,并为未来针对 IV 型柔毛组装 ATP 酶的治疗药物开发提供更好的信息。
Identification of small molecule inhibitors of the Chloracidobacterium thermophilum type IV pilus protein PilB by ensemble virtual screening
Antivirulence strategy has been explored as an alternative to traditional antibiotic development. The bacterial type IV pilus is a virulence factor involved in host invasion and colonization in many antibiotic resistant pathogens. The PilB ATPase hydrolyzes ATP to drive the assembly of the pilus filament from pilin subunits. We evaluated Chloracidobacterium thermophilum PilB (CtPilB) as a model for structure-based virtual screening by molecular docking and molecular dynamics (MD) simulations. A hexameric structure of CtPilB was generated through homology modeling based on an existing crystal structure of a PilB from Geobacter metallireducens. Four representative structures were obtained from molecular dynamics simulations to examine the conformational plasticity of PilB and improve docking analyses by ensemble docking. Structural analyses after 1 μs of simulation revealed conformational changes in individual PilB subunits are dependent on ligand presence. Further, ensemble virtual screening of a library of 4234 compounds retrieved from the ZINC15 database identified five promising PilB inhibitors. Molecular docking and binding analyses using the four representative structures from MD simulations revealed that top-ranked compounds interact with multiple Walker A residues, one Asp-box residue, and one arginine finger, indicating these are key residues in inhibitor binding within the ATP binding pocket. The use of multiple conformations in molecular screening can provide greater insight into compound flexibility within receptor sites and better inform future drug development for therapeutics targeting the type IV pilus assembly ATPase.
期刊介绍:
Archives of Biochemistry and Biophysics publishes quality original articles and reviews in the developing areas of biochemistry and biophysics.
Research Areas Include:
• Enzyme and protein structure, function, regulation. Folding, turnover, and post-translational processing
• Biological oxidations, free radical reactions, redox signaling, oxygenases, P450 reactions
• Signal transduction, receptors, membrane transport, intracellular signals. Cellular and integrated metabolism.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
