Yuanting Zheng , Neil D. Young , Tulio L. Campos , Pasi K. Korhonen , Tao Wang , Sunita B. Sumanam , Aya C. Taki , Joseph J. Byrne , Bill C.H. Chang , Jiangning Song , Robin B. Gasser
{"title":"Haecon-5血吸虫菌株的染色体连续基因组揭示了明显的遗传变异,有助于发现重要的候选基因。","authors":"Yuanting Zheng , Neil D. Young , Tulio L. Campos , Pasi K. Korhonen , Tao Wang , Sunita B. Sumanam , Aya C. Taki , Joseph J. Byrne , Bill C.H. Chang , Jiangning Song , Robin B. Gasser","doi":"10.1016/j.ijpara.2024.08.003","DOIUrl":null,"url":null,"abstract":"<div><div>Millions of livestock animals worldwide are infected with the haematophagous barber’s pole worm, <em>Haemonchus contortus</em>, the aetiological agent of haemonchosis. Despite the major significance of this parasite worldwide and its widespread resistance to current treatments, the lack of a high-quality genome for the well-defined strain of this parasite from Australia, called Haecon-5, has constrained research in a number of areas including host-parasite interactions, drug discovery and population genetics. To enable research in these areas, we report here a chromosome-contiguous genome (∼280 Mb) for Haecon-5 with high-quality models for 19,234 protein-coding genes. Comparative genomic analyses show significant genomic similarity (synteny) with a UK strain of <em>H. contortus</em>, called MHco3(ISE).N1 (abbreviated as “ISE”), but we also discover marked differences in genomic structure/gene arrangements, distribution of nucleotide variability (single nucleotide polymorphisms (SNPs) and indels) and orthology between Haecon-5 and ISE. We used the genome and extensive transcriptomic resources for Haecon-5 to predict a subset of essential single-copy genes employing a “cross-species” machine learning (ML) approach using a range of features from nucleotide/protein sequences, protein orthology, subcellular localisation, single-cell RNA-seq and/or histone methylation data available for the model organisms <em>Caenorhabditis elegans</em> and <em>Drosophila melanogaster</em>. From a set of 1,464 conserved single copy genes, transcribed in key life-cycle stages of <em>H. contortus</em>, we identified 232 genes whose homologs have critical functions in <em>C. elegans</em> and/or <em>D. melanogaster</em>, and prioritised 10 of them for further characterisation; nine of the 10 genes likely play roles in neurophysiological processes, germline, hypodermis and/or respiration, and one is an unknown (orphan) gene for which no detailed functional information exists. Future studies of these genes/gene products are warranted to elucidate their roles in parasite biology, host-parasite interplay and/or disease. Clearly, the present Haecon-5 reference genome and associated resources now underpin a broad range of fundamental investigations of <em>H. contortus</em> and could assist in accelerating the discovery of novel intervention targets and drug candidates to combat haemonchosis.</div></div>","PeriodicalId":13725,"journal":{"name":"International journal for parasitology","volume":"54 13","pages":"Pages 705-715"},"PeriodicalIF":3.2000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Chromosome-contiguous genome for the Haecon-5 strain of Haemonchus contortus reveals marked genetic variability and enables the discovery of essential gene candidates\",\"authors\":\"Yuanting Zheng , Neil D. Young , Tulio L. Campos , Pasi K. Korhonen , Tao Wang , Sunita B. Sumanam , Aya C. Taki , Joseph J. Byrne , Bill C.H. Chang , Jiangning Song , Robin B. Gasser\",\"doi\":\"10.1016/j.ijpara.2024.08.003\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Millions of livestock animals worldwide are infected with the haematophagous barber’s pole worm, <em>Haemonchus contortus</em>, the aetiological agent of haemonchosis. Despite the major significance of this parasite worldwide and its widespread resistance to current treatments, the lack of a high-quality genome for the well-defined strain of this parasite from Australia, called Haecon-5, has constrained research in a number of areas including host-parasite interactions, drug discovery and population genetics. To enable research in these areas, we report here a chromosome-contiguous genome (∼280 Mb) for Haecon-5 with high-quality models for 19,234 protein-coding genes. Comparative genomic analyses show significant genomic similarity (synteny) with a UK strain of <em>H. contortus</em>, called MHco3(ISE).N1 (abbreviated as “ISE”), but we also discover marked differences in genomic structure/gene arrangements, distribution of nucleotide variability (single nucleotide polymorphisms (SNPs) and indels) and orthology between Haecon-5 and ISE. We used the genome and extensive transcriptomic resources for Haecon-5 to predict a subset of essential single-copy genes employing a “cross-species” machine learning (ML) approach using a range of features from nucleotide/protein sequences, protein orthology, subcellular localisation, single-cell RNA-seq and/or histone methylation data available for the model organisms <em>Caenorhabditis elegans</em> and <em>Drosophila melanogaster</em>. From a set of 1,464 conserved single copy genes, transcribed in key life-cycle stages of <em>H. contortus</em>, we identified 232 genes whose homologs have critical functions in <em>C. elegans</em> and/or <em>D. melanogaster</em>, and prioritised 10 of them for further characterisation; nine of the 10 genes likely play roles in neurophysiological processes, germline, hypodermis and/or respiration, and one is an unknown (orphan) gene for which no detailed functional information exists. Future studies of these genes/gene products are warranted to elucidate their roles in parasite biology, host-parasite interplay and/or disease. Clearly, the present Haecon-5 reference genome and associated resources now underpin a broad range of fundamental investigations of <em>H. contortus</em> and could assist in accelerating the discovery of novel intervention targets and drug candidates to combat haemonchosis.</div></div>\",\"PeriodicalId\":13725,\"journal\":{\"name\":\"International journal for parasitology\",\"volume\":\"54 13\",\"pages\":\"Pages 705-715\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"International journal for parasitology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S002075192400153X\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"PARASITOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"International journal for parasitology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S002075192400153X","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/20 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"PARASITOLOGY","Score":null,"Total":0}
Chromosome-contiguous genome for the Haecon-5 strain of Haemonchus contortus reveals marked genetic variability and enables the discovery of essential gene candidates
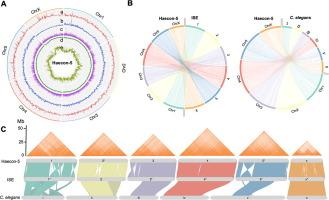
Millions of livestock animals worldwide are infected with the haematophagous barber’s pole worm, Haemonchus contortus, the aetiological agent of haemonchosis. Despite the major significance of this parasite worldwide and its widespread resistance to current treatments, the lack of a high-quality genome for the well-defined strain of this parasite from Australia, called Haecon-5, has constrained research in a number of areas including host-parasite interactions, drug discovery and population genetics. To enable research in these areas, we report here a chromosome-contiguous genome (∼280 Mb) for Haecon-5 with high-quality models for 19,234 protein-coding genes. Comparative genomic analyses show significant genomic similarity (synteny) with a UK strain of H. contortus, called MHco3(ISE).N1 (abbreviated as “ISE”), but we also discover marked differences in genomic structure/gene arrangements, distribution of nucleotide variability (single nucleotide polymorphisms (SNPs) and indels) and orthology between Haecon-5 and ISE. We used the genome and extensive transcriptomic resources for Haecon-5 to predict a subset of essential single-copy genes employing a “cross-species” machine learning (ML) approach using a range of features from nucleotide/protein sequences, protein orthology, subcellular localisation, single-cell RNA-seq and/or histone methylation data available for the model organisms Caenorhabditis elegans and Drosophila melanogaster. From a set of 1,464 conserved single copy genes, transcribed in key life-cycle stages of H. contortus, we identified 232 genes whose homologs have critical functions in C. elegans and/or D. melanogaster, and prioritised 10 of them for further characterisation; nine of the 10 genes likely play roles in neurophysiological processes, germline, hypodermis and/or respiration, and one is an unknown (orphan) gene for which no detailed functional information exists. Future studies of these genes/gene products are warranted to elucidate their roles in parasite biology, host-parasite interplay and/or disease. Clearly, the present Haecon-5 reference genome and associated resources now underpin a broad range of fundamental investigations of H. contortus and could assist in accelerating the discovery of novel intervention targets and drug candidates to combat haemonchosis.
期刊介绍:
International Journal for Parasitology offers authors the option to sponsor nonsubscriber access to their articles on Elsevier electronic publishing platforms. For more information please view our Sponsored Articles page. The International Journal for Parasitology publishes the results of original research in all aspects of basic and applied parasitology, including all the fields covered by its Specialist Editors, and ranging from parasites and host-parasite relationships of intrinsic biological interest to those of social and economic importance in human and veterinary medicine and agriculture.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
