{"title":"对二氧化铪铁电机制的计算理解取得进展","authors":"Tianyuan Zhu, Liyang Ma, Shiqing Deng, Shi Liu","doi":"10.1038/s41524-024-01352-0","DOIUrl":null,"url":null,"abstract":"<p>Since the first report of ferroelectricity in nanoscale HfO<sub>2</sub>-based thin films in 2011, this silicon-compatible binary oxide has quickly garnered intense interest in academia and industry, and continues to do so. Despite its deceivingly simple chemical composition, the ferroelectric physics supported by HfO<sub>2</sub> is remarkably complex, arguably rivaling that of perovskite ferroelectrics. Computational investigations, especially those utilizing first-principles density functional theory (DFT), have significantly advanced our understanding of the nature of ferroelectricity in these thin films. In this review, we provide an in-depth discussion of the computational efforts to understand ferroelectric hafnia, comparing various metastable polar phases and examining the critical factors necessary for their stabilization. The intricate nature of HfO<sub>2</sub> is intimately related to the complex interplay among diverse structural polymorphs, dopants and their charge-compensating oxygen vacancies, and unconventional switching mechanisms of domains and domain walls, which can sometimes yield conflicting theoretical predictions and theoretical-experimental discrepancies. We also discuss opportunities enabled by machine-learning-assisted molecular dynamics and phase-field simulations to go beyond DFT modeling, probing the dynamical properties of ferroelectric HfO<sub>2</sub> and tackling pressing issues such as high coercive fields.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"5 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-08-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Progress in computational understanding of ferroelectric mechanisms in HfO2\",\"authors\":\"Tianyuan Zhu, Liyang Ma, Shiqing Deng, Shi Liu\",\"doi\":\"10.1038/s41524-024-01352-0\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Since the first report of ferroelectricity in nanoscale HfO<sub>2</sub>-based thin films in 2011, this silicon-compatible binary oxide has quickly garnered intense interest in academia and industry, and continues to do so. Despite its deceivingly simple chemical composition, the ferroelectric physics supported by HfO<sub>2</sub> is remarkably complex, arguably rivaling that of perovskite ferroelectrics. Computational investigations, especially those utilizing first-principles density functional theory (DFT), have significantly advanced our understanding of the nature of ferroelectricity in these thin films. In this review, we provide an in-depth discussion of the computational efforts to understand ferroelectric hafnia, comparing various metastable polar phases and examining the critical factors necessary for their stabilization. The intricate nature of HfO<sub>2</sub> is intimately related to the complex interplay among diverse structural polymorphs, dopants and their charge-compensating oxygen vacancies, and unconventional switching mechanisms of domains and domain walls, which can sometimes yield conflicting theoretical predictions and theoretical-experimental discrepancies. We also discuss opportunities enabled by machine-learning-assisted molecular dynamics and phase-field simulations to go beyond DFT modeling, probing the dynamical properties of ferroelectric HfO<sub>2</sub> and tackling pressing issues such as high coercive fields.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"5 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-08-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01352-0\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01352-0","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Progress in computational understanding of ferroelectric mechanisms in HfO2
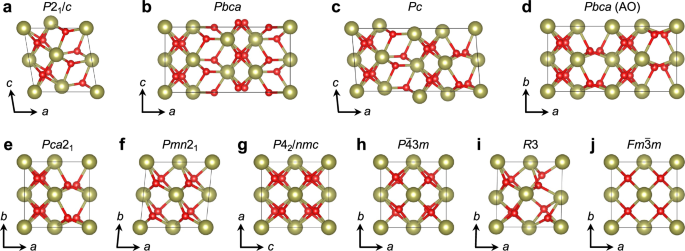
Since the first report of ferroelectricity in nanoscale HfO2-based thin films in 2011, this silicon-compatible binary oxide has quickly garnered intense interest in academia and industry, and continues to do so. Despite its deceivingly simple chemical composition, the ferroelectric physics supported by HfO2 is remarkably complex, arguably rivaling that of perovskite ferroelectrics. Computational investigations, especially those utilizing first-principles density functional theory (DFT), have significantly advanced our understanding of the nature of ferroelectricity in these thin films. In this review, we provide an in-depth discussion of the computational efforts to understand ferroelectric hafnia, comparing various metastable polar phases and examining the critical factors necessary for their stabilization. The intricate nature of HfO2 is intimately related to the complex interplay among diverse structural polymorphs, dopants and their charge-compensating oxygen vacancies, and unconventional switching mechanisms of domains and domain walls, which can sometimes yield conflicting theoretical predictions and theoretical-experimental discrepancies. We also discuss opportunities enabled by machine-learning-assisted molecular dynamics and phase-field simulations to go beyond DFT modeling, probing the dynamical properties of ferroelectric HfO2 and tackling pressing issues such as high coercive fields.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
