Xue Xiao , Tingting Tang , Mingxia Bi , Jing Liu , Mengru Liu , Qian Jiao , Xi Chen , Chunling Yan , Xixun Du , Hong Jiang
{"title":"GHSR 缺乏症通过损害自噬作用加剧帕金森病的病理变化","authors":"Xue Xiao , Tingting Tang , Mingxia Bi , Jing Liu , Mengru Liu , Qian Jiao , Xi Chen , Chunling Yan , Xixun Du , Hong Jiang","doi":"10.1016/j.redox.2024.103322","DOIUrl":null,"url":null,"abstract":"<div><p>In Parkinson's disease (PD), exogenous ghrelin protects dopaminergic neurons through its receptor, growth hormone secretagogue receptor (GHSR). However, in contrast to the strikingly low levels of ghrelin, GHSR is highly expressed in the substantia nigra (SN). What role does GHSR play in dopaminergic neurons is unknown. In this study, using GHSR knockout mice (<em>Ghsr</em><sup><em>−/−</em></sup> mice) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model, we found that GHSR deletion aggravated dopaminergic neurons degeneration, and the expression and activity of GHSR were significantly reduced in PD. Furthermore, we explored the potential mechanism that GHSR deficiency aggregated PD-related neurodegeneration. We showed that DEPTOR, a subunit of mTORC1, was overexpressed in <em>Ghsr</em><sup><em>−/−</em></sup> mice, positively regulating autophagy and enhancing autophagy initiation. The expression of lysosomal markers was abnormal, implying lysosomal dysfunction. As a result, the damaged mitochondria could not be effectively eliminated, which ultimately exacerbated the injury of nigral dopaminergic neurons. In particular, we demonstrated that DEPTOR could be transcriptionally regulated by KLF4. Specific knockdown of KLF4 in dopaminergic neurons effectively alleviated neurodegeneration in <em>Ghsr</em><sup><em>−/−</em></sup> mice. In summary, our results suggested that endogenous GHSR deletion-compromised autophagy by impairing lysosomal function, is a key contributor to PD, which provided ideas for therapeutic approaches involving the manipulation of GHSR.</p></div>","PeriodicalId":20998,"journal":{"name":"Redox Biology","volume":"76 ","pages":"Article 103322"},"PeriodicalIF":11.9000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2213231724003008/pdfft?md5=2feba14bab097032a2f68c401ca25097&pid=1-s2.0-S2213231724003008-main.pdf","citationCount":"0","resultStr":"{\"title\":\"GHSR deficiency exacerbates Parkinson's disease pathology by impairing autophagy\",\"authors\":\"Xue Xiao , Tingting Tang , Mingxia Bi , Jing Liu , Mengru Liu , Qian Jiao , Xi Chen , Chunling Yan , Xixun Du , Hong Jiang\",\"doi\":\"10.1016/j.redox.2024.103322\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><p>In Parkinson's disease (PD), exogenous ghrelin protects dopaminergic neurons through its receptor, growth hormone secretagogue receptor (GHSR). However, in contrast to the strikingly low levels of ghrelin, GHSR is highly expressed in the substantia nigra (SN). What role does GHSR play in dopaminergic neurons is unknown. In this study, using GHSR knockout mice (<em>Ghsr</em><sup><em>−/−</em></sup> mice) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model, we found that GHSR deletion aggravated dopaminergic neurons degeneration, and the expression and activity of GHSR were significantly reduced in PD. Furthermore, we explored the potential mechanism that GHSR deficiency aggregated PD-related neurodegeneration. We showed that DEPTOR, a subunit of mTORC1, was overexpressed in <em>Ghsr</em><sup><em>−/−</em></sup> mice, positively regulating autophagy and enhancing autophagy initiation. The expression of lysosomal markers was abnormal, implying lysosomal dysfunction. As a result, the damaged mitochondria could not be effectively eliminated, which ultimately exacerbated the injury of nigral dopaminergic neurons. In particular, we demonstrated that DEPTOR could be transcriptionally regulated by KLF4. Specific knockdown of KLF4 in dopaminergic neurons effectively alleviated neurodegeneration in <em>Ghsr</em><sup><em>−/−</em></sup> mice. In summary, our results suggested that endogenous GHSR deletion-compromised autophagy by impairing lysosomal function, is a key contributor to PD, which provided ideas for therapeutic approaches involving the manipulation of GHSR.</p></div>\",\"PeriodicalId\":20998,\"journal\":{\"name\":\"Redox Biology\",\"volume\":\"76 \",\"pages\":\"Article 103322\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2213231724003008/pdfft?md5=2feba14bab097032a2f68c401ca25097&pid=1-s2.0-S2213231724003008-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Redox Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2213231724003008\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/20 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Redox Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2213231724003008","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/20 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
GHSR deficiency exacerbates Parkinson's disease pathology by impairing autophagy
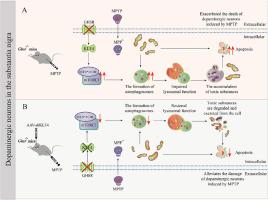
In Parkinson's disease (PD), exogenous ghrelin protects dopaminergic neurons through its receptor, growth hormone secretagogue receptor (GHSR). However, in contrast to the strikingly low levels of ghrelin, GHSR is highly expressed in the substantia nigra (SN). What role does GHSR play in dopaminergic neurons is unknown. In this study, using GHSR knockout mice (Ghsr−/− mice) and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD model, we found that GHSR deletion aggravated dopaminergic neurons degeneration, and the expression and activity of GHSR were significantly reduced in PD. Furthermore, we explored the potential mechanism that GHSR deficiency aggregated PD-related neurodegeneration. We showed that DEPTOR, a subunit of mTORC1, was overexpressed in Ghsr−/− mice, positively regulating autophagy and enhancing autophagy initiation. The expression of lysosomal markers was abnormal, implying lysosomal dysfunction. As a result, the damaged mitochondria could not be effectively eliminated, which ultimately exacerbated the injury of nigral dopaminergic neurons. In particular, we demonstrated that DEPTOR could be transcriptionally regulated by KLF4. Specific knockdown of KLF4 in dopaminergic neurons effectively alleviated neurodegeneration in Ghsr−/− mice. In summary, our results suggested that endogenous GHSR deletion-compromised autophagy by impairing lysosomal function, is a key contributor to PD, which provided ideas for therapeutic approaches involving the manipulation of GHSR.
期刊介绍:
Redox Biology is the official journal of the Society for Redox Biology and Medicine and the Society for Free Radical Research-Europe. It is also affiliated with the International Society for Free Radical Research (SFRRI). This journal serves as a platform for publishing pioneering research, innovative methods, and comprehensive review articles in the field of redox biology, encompassing both health and disease.
Redox Biology welcomes various forms of contributions, including research articles (short or full communications), methods, mini-reviews, and commentaries. Through its diverse range of published content, Redox Biology aims to foster advancements and insights in the understanding of redox biology and its implications.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
