{"title":"多 GPU RI-HF 能量和分析梯度--面向高通量 Ab Initio 分子动力学。","authors":"Ryan Stocks, Elise Palethorpe, Giuseppe M J Barca","doi":"10.1021/acs.jctc.4c00877","DOIUrl":null,"url":null,"abstract":"<p><p>This article presents an optimized algorithm and implementation for calculating resolution-of-the-identity Hartree-Fock (RI-HF) energies and analytic gradients using multiple graphics processing units (GPUs). The algorithm is especially designed for high throughput ab initio molecular dynamics simulations of small and medium size molecules (10-100 atoms). Key innovations of this work include the exploitation of multi-GPU parallelism and a workload balancing scheme that efficiently distributes computational tasks among GPUs. Our implementation also employs techniques for symmetry utilization, integral screening, and leveraging sparsity to optimize memory usage. Computational results show that the implementation achieves significant performance improvements, including over 3 × speedups in single GPU AIMD throughput compared to previous GPU-accelerated RI-HF and traditional HF methods. Furthermore, utilizing multiple GPUs can provide superlinear speedup when the additional aggregate GPU memory allows for the storage of decompressed three-center integrals.</p>","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":" ","pages":"7503-7515"},"PeriodicalIF":5.8000,"publicationDate":"2024-09-10","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Multi-GPU RI-HF Energies and Analytic Gradients─Toward High-Throughput Ab Initio Molecular Dynamics.\",\"authors\":\"Ryan Stocks, Elise Palethorpe, Giuseppe M J Barca\",\"doi\":\"10.1021/acs.jctc.4c00877\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>This article presents an optimized algorithm and implementation for calculating resolution-of-the-identity Hartree-Fock (RI-HF) energies and analytic gradients using multiple graphics processing units (GPUs). The algorithm is especially designed for high throughput ab initio molecular dynamics simulations of small and medium size molecules (10-100 atoms). Key innovations of this work include the exploitation of multi-GPU parallelism and a workload balancing scheme that efficiently distributes computational tasks among GPUs. Our implementation also employs techniques for symmetry utilization, integral screening, and leveraging sparsity to optimize memory usage. Computational results show that the implementation achieves significant performance improvements, including over 3 × speedups in single GPU AIMD throughput compared to previous GPU-accelerated RI-HF and traditional HF methods. Furthermore, utilizing multiple GPUs can provide superlinear speedup when the additional aggregate GPU memory allows for the storage of decompressed three-center integrals.</p>\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\" \",\"pages\":\"7503-7515\"},\"PeriodicalIF\":5.8000,\"publicationDate\":\"2024-09-10\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00877\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/8/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00877","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/8/27 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Multi-GPU RI-HF Energies and Analytic Gradients─Toward High-Throughput Ab Initio Molecular Dynamics.
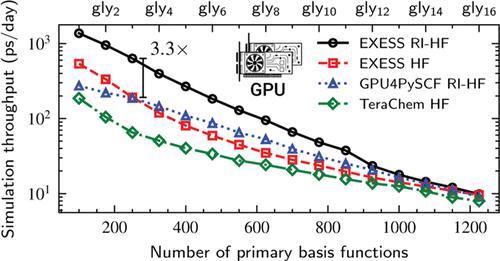
This article presents an optimized algorithm and implementation for calculating resolution-of-the-identity Hartree-Fock (RI-HF) energies and analytic gradients using multiple graphics processing units (GPUs). The algorithm is especially designed for high throughput ab initio molecular dynamics simulations of small and medium size molecules (10-100 atoms). Key innovations of this work include the exploitation of multi-GPU parallelism and a workload balancing scheme that efficiently distributes computational tasks among GPUs. Our implementation also employs techniques for symmetry utilization, integral screening, and leveraging sparsity to optimize memory usage. Computational results show that the implementation achieves significant performance improvements, including over 3 × speedups in single GPU AIMD throughput compared to previous GPU-accelerated RI-HF and traditional HF methods. Furthermore, utilizing multiple GPUs can provide superlinear speedup when the additional aggregate GPU memory allows for the storage of decompressed three-center integrals.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
