{"title":"GRK2- 介导的 AKT 激活以 p53 依赖性方式控制细胞周期进展和 G2 检查点。","authors":"Verónica Rivas, Teresa González-Muñoz, Ángela Albitre, Vanesa Lafarga, Cristina Delgado-Arévalo, Federico Mayor, Petronila Penela","doi":"10.1038/s41420-024-02143-8","DOIUrl":null,"url":null,"abstract":"<p><p>Cell cycle checkpoints, activated by stressful events, halt the cell cycle progression, and prevent the transmission of damaged DNA. These checkpoints prompt cell repair but also trigger cell death if damage persists. Decision-making between these responses is multifactorial and context-dependent, with the tumor suppressor p53 playing a central role. In many tumor cells, p53 alterations lead to G1/S checkpoint loss and the weakening of the G2 checkpoint, rendering cell viability dependent on the strength of the latter through mechanisms not fully characterized. Cells with a strong pro-survival drive can evade cell death despite substantial DNA lesions. Deciphering the integration of survival pathways with p53-dependent and -independent mechanisms governing the G2/M transition is crucial for understanding G2 arrest functionality and predicting tumor cell response to chemotherapy. The serine/threonine kinase GRK2 emerges as a signaling node in cell cycle modulation. In cycling cells, but not in G2 checkpoint-arrested cells, GRK2 protein levels decline during G2/M transition through a process triggered by CDK2-dependent phosphorylation of GRK2 at the S670 residue and Mdm2 ubiquitination. We report now that this downmodulation in G2 prevents the unscheduled activation of the PI3K/AKT pathway, allowing cells to progress into mitosis. Conversely, higher GRK2 levels lead to tyrosine phosphorylation by the kinase c-Abl, promoting the direct association of GRK2 with the p85 regulatory subunit of PI3K and AKT activation in a GRK2 catalytic-independent manner. Hyperactivation of AKT is conditioned by p53's scaffolding function, triggering FOXO3a phosphorylation, impaired Cyclin B1 accumulation, and CDK1 activation, causing a G2/M transition delay. Upon G2 checkpoint activation, GRK2 potentiates early arrest independently of p53 through AKT activation. However, its ability to overcome the G2 checkpoint in viable conditions depends on p53. Our results suggest that integrating the GRK2/PI3K/AKT axis with non-canonical functions of p53 might confer a survival advantage to tumor cells.</p>","PeriodicalId":9735,"journal":{"name":"Cell Death Discovery","volume":"10 1","pages":"385"},"PeriodicalIF":7.0000,"publicationDate":"2024-08-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11358448/pdf/","citationCount":"0","resultStr":"{\"title\":\"GRK2-mediated AKT activation controls cell cycle progression and G2 checkpoint in a p53-dependent manner.\",\"authors\":\"Verónica Rivas, Teresa González-Muñoz, Ángela Albitre, Vanesa Lafarga, Cristina Delgado-Arévalo, Federico Mayor, Petronila Penela\",\"doi\":\"10.1038/s41420-024-02143-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Cell cycle checkpoints, activated by stressful events, halt the cell cycle progression, and prevent the transmission of damaged DNA. These checkpoints prompt cell repair but also trigger cell death if damage persists. Decision-making between these responses is multifactorial and context-dependent, with the tumor suppressor p53 playing a central role. In many tumor cells, p53 alterations lead to G1/S checkpoint loss and the weakening of the G2 checkpoint, rendering cell viability dependent on the strength of the latter through mechanisms not fully characterized. Cells with a strong pro-survival drive can evade cell death despite substantial DNA lesions. Deciphering the integration of survival pathways with p53-dependent and -independent mechanisms governing the G2/M transition is crucial for understanding G2 arrest functionality and predicting tumor cell response to chemotherapy. The serine/threonine kinase GRK2 emerges as a signaling node in cell cycle modulation. In cycling cells, but not in G2 checkpoint-arrested cells, GRK2 protein levels decline during G2/M transition through a process triggered by CDK2-dependent phosphorylation of GRK2 at the S670 residue and Mdm2 ubiquitination. We report now that this downmodulation in G2 prevents the unscheduled activation of the PI3K/AKT pathway, allowing cells to progress into mitosis. Conversely, higher GRK2 levels lead to tyrosine phosphorylation by the kinase c-Abl, promoting the direct association of GRK2 with the p85 regulatory subunit of PI3K and AKT activation in a GRK2 catalytic-independent manner. Hyperactivation of AKT is conditioned by p53's scaffolding function, triggering FOXO3a phosphorylation, impaired Cyclin B1 accumulation, and CDK1 activation, causing a G2/M transition delay. Upon G2 checkpoint activation, GRK2 potentiates early arrest independently of p53 through AKT activation. However, its ability to overcome the G2 checkpoint in viable conditions depends on p53. Our results suggest that integrating the GRK2/PI3K/AKT axis with non-canonical functions of p53 might confer a survival advantage to tumor cells.</p>\",\"PeriodicalId\":9735,\"journal\":{\"name\":\"Cell Death Discovery\",\"volume\":\"10 1\",\"pages\":\"385\"},\"PeriodicalIF\":7.0000,\"publicationDate\":\"2024-08-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11358448/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cell Death Discovery\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41420-024-02143-8\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death Discovery","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41420-024-02143-8","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
GRK2-mediated AKT activation controls cell cycle progression and G2 checkpoint in a p53-dependent manner.
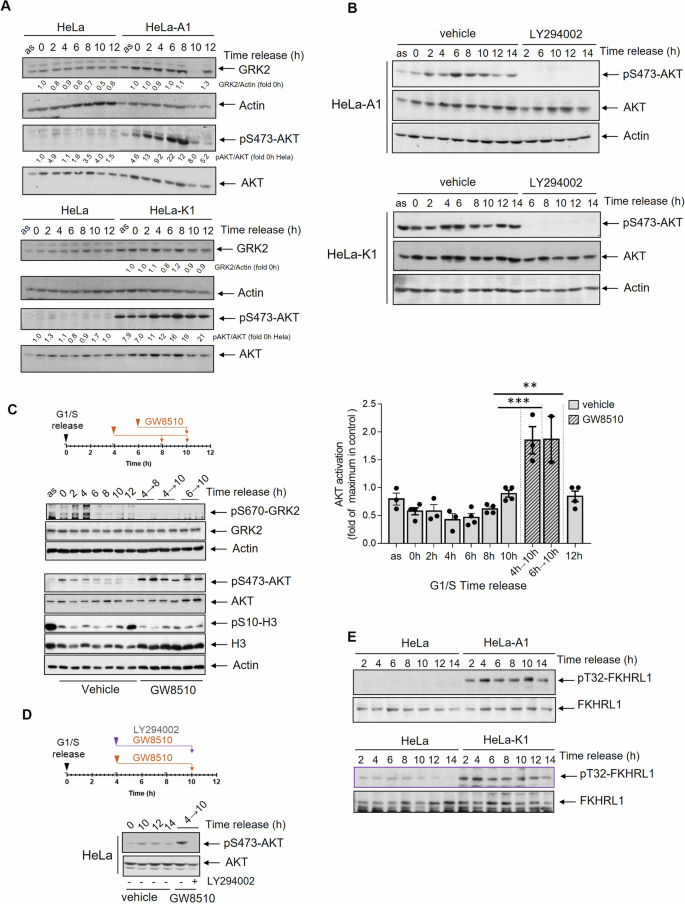
Cell cycle checkpoints, activated by stressful events, halt the cell cycle progression, and prevent the transmission of damaged DNA. These checkpoints prompt cell repair but also trigger cell death if damage persists. Decision-making between these responses is multifactorial and context-dependent, with the tumor suppressor p53 playing a central role. In many tumor cells, p53 alterations lead to G1/S checkpoint loss and the weakening of the G2 checkpoint, rendering cell viability dependent on the strength of the latter through mechanisms not fully characterized. Cells with a strong pro-survival drive can evade cell death despite substantial DNA lesions. Deciphering the integration of survival pathways with p53-dependent and -independent mechanisms governing the G2/M transition is crucial for understanding G2 arrest functionality and predicting tumor cell response to chemotherapy. The serine/threonine kinase GRK2 emerges as a signaling node in cell cycle modulation. In cycling cells, but not in G2 checkpoint-arrested cells, GRK2 protein levels decline during G2/M transition through a process triggered by CDK2-dependent phosphorylation of GRK2 at the S670 residue and Mdm2 ubiquitination. We report now that this downmodulation in G2 prevents the unscheduled activation of the PI3K/AKT pathway, allowing cells to progress into mitosis. Conversely, higher GRK2 levels lead to tyrosine phosphorylation by the kinase c-Abl, promoting the direct association of GRK2 with the p85 regulatory subunit of PI3K and AKT activation in a GRK2 catalytic-independent manner. Hyperactivation of AKT is conditioned by p53's scaffolding function, triggering FOXO3a phosphorylation, impaired Cyclin B1 accumulation, and CDK1 activation, causing a G2/M transition delay. Upon G2 checkpoint activation, GRK2 potentiates early arrest independently of p53 through AKT activation. However, its ability to overcome the G2 checkpoint in viable conditions depends on p53. Our results suggest that integrating the GRK2/PI3K/AKT axis with non-canonical functions of p53 might confer a survival advantage to tumor cells.
期刊介绍:
Cell Death Discovery is a multidisciplinary, international, online-only, open access journal, dedicated to publishing research at the intersection of medicine with biochemistry, pharmacology, immunology, cell biology and cell death, provided it is scientifically sound. The unrestricted access to research findings in Cell Death Discovery will foster a dynamic and highly productive dialogue between basic scientists and clinicians, as well as researchers in industry with a focus on cancer, neurobiology and inflammation research. As an official journal of the Cell Death Differentiation Association (ADMC), Cell Death Discovery will build upon the success of Cell Death & Differentiation and Cell Death & Disease in publishing important peer-reviewed original research, timely reviews and editorial commentary.
Cell Death Discovery is committed to increasing the reproducibility of research. To this end, in conjunction with its sister journals Cell Death & Differentiation and Cell Death & Disease, Cell Death Discovery provides a unique forum for scientists as well as clinicians and members of the pharmaceutical and biotechnical industry. It is committed to the rapid publication of high quality original papers that relate to these subjects, together with topical, usually solicited, reviews, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
