Matheus Máximo-Canadas, Lucas Modesto-Costa, Itamar Borges Jr
{"title":"对硝基苯胺在不同溶剂中的 Ab initio 电子吸收光谱:分子内电荷转移效应","authors":"Matheus Máximo-Canadas, Lucas Modesto-Costa, Itamar Borges Jr","doi":"10.1002/jcc.27493","DOIUrl":null,"url":null,"abstract":"<p>Intramolecular charge transfer (ICT) effects of <i>para</i>-nitroaniline (<i>p</i>NA) in eight solvents (cyclohexane, toluene, acetic acid, dichloroethane, acetone, acetonitrile, dimethylsulfoxide, and water) are investigated extensively. The second-order algebraic diagrammatic construction, ADC(2), ab initio wave function is employed with the COSMO implicit and discrete multiscale solvation methods. We found a decreasing amine group torsion angle with increased solvent polarity and a linear correlation between the polarity and ADC(2) transition energies. The first absorption band involves π → π* transitions with ICT from the amine and the benzene ring to the nitro group, increased by 4%–11% for different solvation models of water compared to the vacuum. A second band of <i>p</i>NA is characterized for the first time. This band is primarily a local excitation on the nitro group, including some ICT from the amine group to the benzene ring that decreases with the solvent polarity. For cyclohexane, the COSMO implicit solvent model shows the best agreement with the experiment, while the explicit model has the best agreement for water.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"45 32","pages":"2899-2911"},"PeriodicalIF":4.8000,"publicationDate":"2024-08-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Ab initio electronic absorption spectra of para-nitroaniline in different solvents: Intramolecular charge transfer effects\",\"authors\":\"Matheus Máximo-Canadas, Lucas Modesto-Costa, Itamar Borges Jr\",\"doi\":\"10.1002/jcc.27493\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Intramolecular charge transfer (ICT) effects of <i>para</i>-nitroaniline (<i>p</i>NA) in eight solvents (cyclohexane, toluene, acetic acid, dichloroethane, acetone, acetonitrile, dimethylsulfoxide, and water) are investigated extensively. The second-order algebraic diagrammatic construction, ADC(2), ab initio wave function is employed with the COSMO implicit and discrete multiscale solvation methods. We found a decreasing amine group torsion angle with increased solvent polarity and a linear correlation between the polarity and ADC(2) transition energies. The first absorption band involves π → π* transitions with ICT from the amine and the benzene ring to the nitro group, increased by 4%–11% for different solvation models of water compared to the vacuum. A second band of <i>p</i>NA is characterized for the first time. This band is primarily a local excitation on the nitro group, including some ICT from the amine group to the benzene ring that decreases with the solvent polarity. For cyclohexane, the COSMO implicit solvent model shows the best agreement with the experiment, while the explicit model has the best agreement for water.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"45 32\",\"pages\":\"2899-2911\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-08-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27493\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27493","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Ab initio electronic absorption spectra of para-nitroaniline in different solvents: Intramolecular charge transfer effects
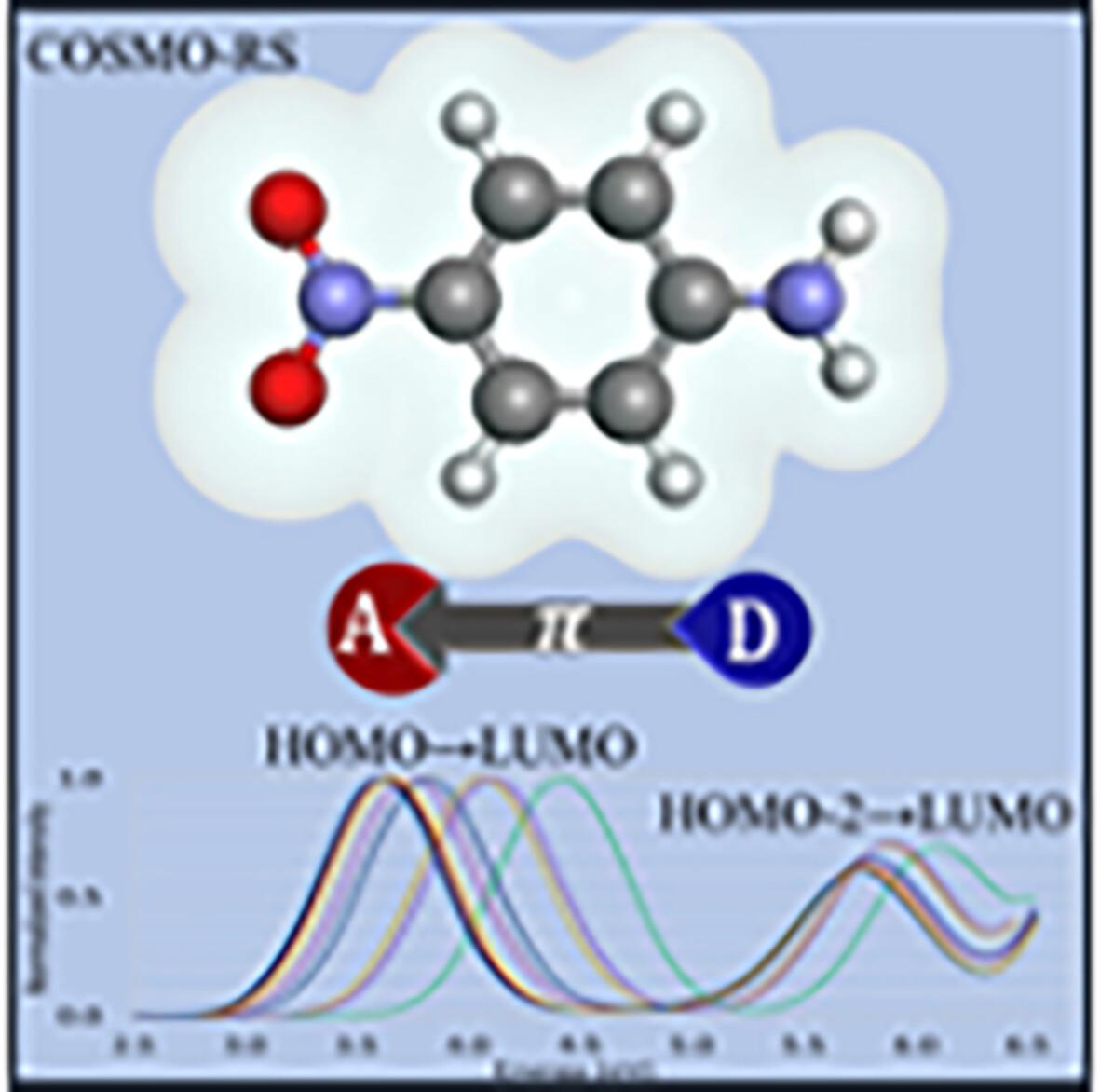
Intramolecular charge transfer (ICT) effects of para-nitroaniline (pNA) in eight solvents (cyclohexane, toluene, acetic acid, dichloroethane, acetone, acetonitrile, dimethylsulfoxide, and water) are investigated extensively. The second-order algebraic diagrammatic construction, ADC(2), ab initio wave function is employed with the COSMO implicit and discrete multiscale solvation methods. We found a decreasing amine group torsion angle with increased solvent polarity and a linear correlation between the polarity and ADC(2) transition energies. The first absorption band involves π → π* transitions with ICT from the amine and the benzene ring to the nitro group, increased by 4%–11% for different solvation models of water compared to the vacuum. A second band of pNA is characterized for the first time. This band is primarily a local excitation on the nitro group, including some ICT from the amine group to the benzene ring that decreases with the solvent polarity. For cyclohexane, the COSMO implicit solvent model shows the best agreement with the experiment, while the explicit model has the best agreement for water.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
