Yu-Shi Liu, Wen-Shuo Yuan, Qi-Jun Liu, Fu-Sheng Liu, Zheng-Tang Liu
{"title":"新型高能离子 5,5'-二硝基氨基-3,3'-偶氮-恶二唑-4,7-二氨基哒嗪[4,5-c] 呋喃盐的结构、机械、电子、振动特性和氢键。","authors":"Yu-Shi Liu, Wen-Shuo Yuan, Qi-Jun Liu, Fu-Sheng Liu, Zheng-Tang Liu","doi":"10.1007/s00894-024-06124-7","DOIUrl":null,"url":null,"abstract":"<div><h3>Context and results</h3><p>The structure, mechanical, electronic, vibration, and hydrogen bonding properties of a novel high-energy and low-sensitivity 5, 5′-dinitroamino-3, 3′-azo-oxadiazole 4, 7-diaminopyridazino [4, 5-c] furoxan salt have been studied by density functional theory. The calculated vibrational properties show that the low-frequency mode is mainly contributed by the vibration of the -NO2 group, and the high-frequency mode is mainly contributed by the vibration of the -NH2 group and the N7-H3 bond which protonates the cation. In addition, it is analyzed that the first bond to break may be the N-NO2 bond. The calculated hydrogen bond properties indicate that the hydrogen bond between water molecules and cations is N7-H3… O5 (1.563 Å), which is the shortest hydrogen bond among all hydrogen bonds. The presence of this exceptionally short hydrogen bond renders the N7-H3 and H6-O5 bonds resistant to disruption at high frequencies, underscoring the pivotal role of hydrogen bonding in stabilizing the structure of energetic materials. Given the absence of experimental and theoretical data on the electronic, mechanical, and vibrational properties of the material thus far, our calculations offer valuable theoretical insights into the ionic salts of high energy and low sensitivity.</p><h3>Computational methods</h3><p>All calculations have been carried out based on density functional theory (DFT) and implemented in the CASTEP code. The mode-conserving pseudopotential is utilized to describe the plane wave expansion function, while the PBE functional within the generalized gradient approximation (GGA) is employed to characterize the exchange–correlation interaction. Additionally, dispersion correction is applied using Grimme’s DFT-D method.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Structural, mechanical, electronic, vibrational properties and hydrogen bonding of a novel energetic ionic 5, 5′-dinitroamino-3, 3′-azo-oxadiazole 4, 7-diaminopyridazino [4, 5-c] furoxan salt\",\"authors\":\"Yu-Shi Liu, Wen-Shuo Yuan, Qi-Jun Liu, Fu-Sheng Liu, Zheng-Tang Liu\",\"doi\":\"10.1007/s00894-024-06124-7\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context and results</h3><p>The structure, mechanical, electronic, vibration, and hydrogen bonding properties of a novel high-energy and low-sensitivity 5, 5′-dinitroamino-3, 3′-azo-oxadiazole 4, 7-diaminopyridazino [4, 5-c] furoxan salt have been studied by density functional theory. The calculated vibrational properties show that the low-frequency mode is mainly contributed by the vibration of the -NO2 group, and the high-frequency mode is mainly contributed by the vibration of the -NH2 group and the N7-H3 bond which protonates the cation. In addition, it is analyzed that the first bond to break may be the N-NO2 bond. The calculated hydrogen bond properties indicate that the hydrogen bond between water molecules and cations is N7-H3… O5 (1.563 Å), which is the shortest hydrogen bond among all hydrogen bonds. The presence of this exceptionally short hydrogen bond renders the N7-H3 and H6-O5 bonds resistant to disruption at high frequencies, underscoring the pivotal role of hydrogen bonding in stabilizing the structure of energetic materials. Given the absence of experimental and theoretical data on the electronic, mechanical, and vibrational properties of the material thus far, our calculations offer valuable theoretical insights into the ionic salts of high energy and low sensitivity.</p><h3>Computational methods</h3><p>All calculations have been carried out based on density functional theory (DFT) and implemented in the CASTEP code. The mode-conserving pseudopotential is utilized to describe the plane wave expansion function, while the PBE functional within the generalized gradient approximation (GGA) is employed to characterize the exchange–correlation interaction. Additionally, dispersion correction is applied using Grimme’s DFT-D method.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 10\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06124-7\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06124-7","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Structural, mechanical, electronic, vibrational properties and hydrogen bonding of a novel energetic ionic 5, 5′-dinitroamino-3, 3′-azo-oxadiazole 4, 7-diaminopyridazino [4, 5-c] furoxan salt
Context and results
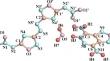
The structure, mechanical, electronic, vibration, and hydrogen bonding properties of a novel high-energy and low-sensitivity 5, 5′-dinitroamino-3, 3′-azo-oxadiazole 4, 7-diaminopyridazino [4, 5-c] furoxan salt have been studied by density functional theory. The calculated vibrational properties show that the low-frequency mode is mainly contributed by the vibration of the -NO2 group, and the high-frequency mode is mainly contributed by the vibration of the -NH2 group and the N7-H3 bond which protonates the cation. In addition, it is analyzed that the first bond to break may be the N-NO2 bond. The calculated hydrogen bond properties indicate that the hydrogen bond between water molecules and cations is N7-H3… O5 (1.563 Å), which is the shortest hydrogen bond among all hydrogen bonds. The presence of this exceptionally short hydrogen bond renders the N7-H3 and H6-O5 bonds resistant to disruption at high frequencies, underscoring the pivotal role of hydrogen bonding in stabilizing the structure of energetic materials. Given the absence of experimental and theoretical data on the electronic, mechanical, and vibrational properties of the material thus far, our calculations offer valuable theoretical insights into the ionic salts of high energy and low sensitivity.
Computational methods
All calculations have been carried out based on density functional theory (DFT) and implemented in the CASTEP code. The mode-conserving pseudopotential is utilized to describe the plane wave expansion function, while the PBE functional within the generalized gradient approximation (GGA) is employed to characterize the exchange–correlation interaction. Additionally, dispersion correction is applied using Grimme’s DFT-D method.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
