{"title":"基于密码子用法和表达的特征大大提高了对 CRISPR 效率的预测。","authors":"Shaked Bergman, Tamir Tuller","doi":"10.1038/s41540-024-00431-8","DOIUrl":null,"url":null,"abstract":"<p><p>CRISPR is a precise and effective genome editing technology; but despite several advancements during the last decade, our ability to computationally design gRNAs remains limited. Most predictive models have relatively low predictive power and utilize only the sequence of the target site as input. Here we suggest a new category of features, which incorporate the target site genomic position and the presence of genes close to it. We calculate four features based on gene expression and codon usage bias indices. We show, on CRISPR datasets taken from 3 different cell types, that such features perform comparably with 425 state-of-the-art predictive features, ranking in the top 2-12% of features. We trained new predictive models, showing that adding expression features to them significantly improves their r<sup>2</sup> by up to 0.04 (relative increase of 39%), achieving average correlations of up to 0.38 on their validation sets; and that these features are deemed important by different feature importance metrics. We believe that incorporating the target site's position, in addition to its sequence, in features such as we have generated here will improve our ability to predict, design and understand CRISPR experiments going forward.</p>","PeriodicalId":19345,"journal":{"name":"NPJ Systems Biology and Applications","volume":"10 1","pages":"100"},"PeriodicalIF":3.5000,"publicationDate":"2024-09-03","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11372048/pdf/","citationCount":"0","resultStr":"{\"title\":\"Codon usage and expression-based features significantly improve prediction of CRISPR efficiency.\",\"authors\":\"Shaked Bergman, Tamir Tuller\",\"doi\":\"10.1038/s41540-024-00431-8\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>CRISPR is a precise and effective genome editing technology; but despite several advancements during the last decade, our ability to computationally design gRNAs remains limited. Most predictive models have relatively low predictive power and utilize only the sequence of the target site as input. Here we suggest a new category of features, which incorporate the target site genomic position and the presence of genes close to it. We calculate four features based on gene expression and codon usage bias indices. We show, on CRISPR datasets taken from 3 different cell types, that such features perform comparably with 425 state-of-the-art predictive features, ranking in the top 2-12% of features. We trained new predictive models, showing that adding expression features to them significantly improves their r<sup>2</sup> by up to 0.04 (relative increase of 39%), achieving average correlations of up to 0.38 on their validation sets; and that these features are deemed important by different feature importance metrics. We believe that incorporating the target site's position, in addition to its sequence, in features such as we have generated here will improve our ability to predict, design and understand CRISPR experiments going forward.</p>\",\"PeriodicalId\":19345,\"journal\":{\"name\":\"NPJ Systems Biology and Applications\",\"volume\":\"10 1\",\"pages\":\"100\"},\"PeriodicalIF\":3.5000,\"publicationDate\":\"2024-09-03\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11372048/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Systems Biology and Applications\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1038/s41540-024-00431-8\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"MATHEMATICAL & COMPUTATIONAL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Systems Biology and Applications","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1038/s41540-024-00431-8","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"MATHEMATICAL & COMPUTATIONAL BIOLOGY","Score":null,"Total":0}
Codon usage and expression-based features significantly improve prediction of CRISPR efficiency.
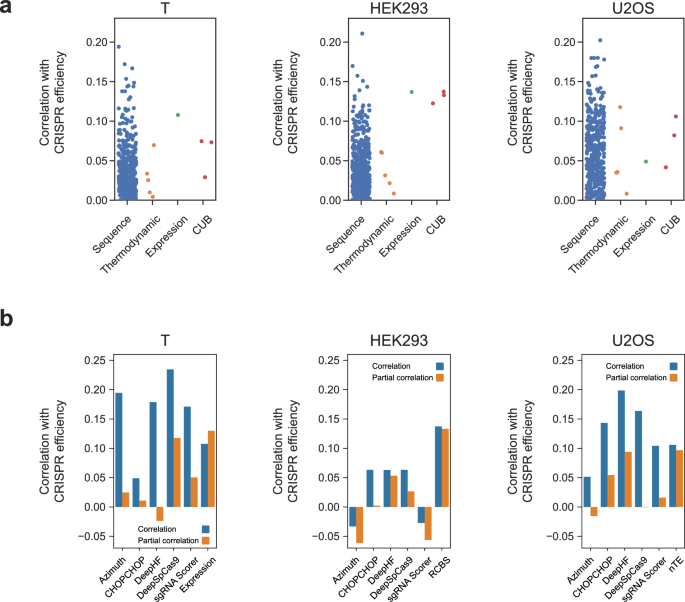
CRISPR is a precise and effective genome editing technology; but despite several advancements during the last decade, our ability to computationally design gRNAs remains limited. Most predictive models have relatively low predictive power and utilize only the sequence of the target site as input. Here we suggest a new category of features, which incorporate the target site genomic position and the presence of genes close to it. We calculate four features based on gene expression and codon usage bias indices. We show, on CRISPR datasets taken from 3 different cell types, that such features perform comparably with 425 state-of-the-art predictive features, ranking in the top 2-12% of features. We trained new predictive models, showing that adding expression features to them significantly improves their r2 by up to 0.04 (relative increase of 39%), achieving average correlations of up to 0.38 on their validation sets; and that these features are deemed important by different feature importance metrics. We believe that incorporating the target site's position, in addition to its sequence, in features such as we have generated here will improve our ability to predict, design and understand CRISPR experiments going forward.
期刊介绍:
npj Systems Biology and Applications is an online Open Access journal dedicated to publishing the premier research that takes a systems-oriented approach. The journal aims to provide a forum for the presentation of articles that help define this nascent field, as well as those that apply the advances to wider fields. We encourage studies that integrate, or aid the integration of, data, analyses and insight from molecules to organisms and broader systems. Important areas of interest include not only fundamental biological systems and drug discovery, but also applications to health, medical practice and implementation, big data, biotechnology, food science, human behaviour, broader biological systems and industrial applications of systems biology.
We encourage all approaches, including network biology, application of control theory to biological systems, computational modelling and analysis, comprehensive and/or high-content measurements, theoretical, analytical and computational studies of system-level properties of biological systems and computational/software/data platforms enabling such studies.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
