{"title":"杂原子和化学功能化对有机半导体晶体结构和载流子迁移率的影响","authors":"S. Hutsch, F. Ortmann","doi":"10.1038/s41524-024-01397-1","DOIUrl":null,"url":null,"abstract":"<p>The substitution of heteroatoms and the functionalisation of molecules are established strategies in chemical synthesis. They target the precise tuning of the electronic properties of hydrocarbon molecules to improve their performance in various applications and increase their versatility. Modifications to the molecular structure often lead to simultaneous changes in the morphology such as different crystal structures. These changes can have a stronger and unpredictable impact on the targeted property. The complex relationships between substitution/functionalization in chemical synthesis and the resulting modifications of properties in thin films or crystals are difficult to predict and remain elusive. Here we address these effects for charge carrier transport in organic crystals by combining simulations of carrier mobilities with crystal structure prediction based on density functional theory and density functional tight binding theory. This enables the prediction of carrier mobilities based solely on the molecular structure and allows for the investigation of chemical modifications prior to synthesis and characterisation. Studying nine specific molecules with tetracene and rubrene as reference compounds along with their combined modifications of the molecular cores and additional functionalisations, we unveil systematic trends for the carrier mobilities of their polymorphs. The positive effect of phenyl groups that is responsible for the marked differences between tetracene and rubrene can be transferred to other small molecules such as NDT and NBT leading to a mobility increase by large factors of about five.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"23 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-09-04","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Impact of heteroatoms and chemical functionalisation on crystal structure and carrier mobility of organic semiconductors\",\"authors\":\"S. Hutsch, F. Ortmann\",\"doi\":\"10.1038/s41524-024-01397-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The substitution of heteroatoms and the functionalisation of molecules are established strategies in chemical synthesis. They target the precise tuning of the electronic properties of hydrocarbon molecules to improve their performance in various applications and increase their versatility. Modifications to the molecular structure often lead to simultaneous changes in the morphology such as different crystal structures. These changes can have a stronger and unpredictable impact on the targeted property. The complex relationships between substitution/functionalization in chemical synthesis and the resulting modifications of properties in thin films or crystals are difficult to predict and remain elusive. Here we address these effects for charge carrier transport in organic crystals by combining simulations of carrier mobilities with crystal structure prediction based on density functional theory and density functional tight binding theory. This enables the prediction of carrier mobilities based solely on the molecular structure and allows for the investigation of chemical modifications prior to synthesis and characterisation. Studying nine specific molecules with tetracene and rubrene as reference compounds along with their combined modifications of the molecular cores and additional functionalisations, we unveil systematic trends for the carrier mobilities of their polymorphs. The positive effect of phenyl groups that is responsible for the marked differences between tetracene and rubrene can be transferred to other small molecules such as NDT and NBT leading to a mobility increase by large factors of about five.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"23 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-09-04\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01397-1\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01397-1","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Impact of heteroatoms and chemical functionalisation on crystal structure and carrier mobility of organic semiconductors
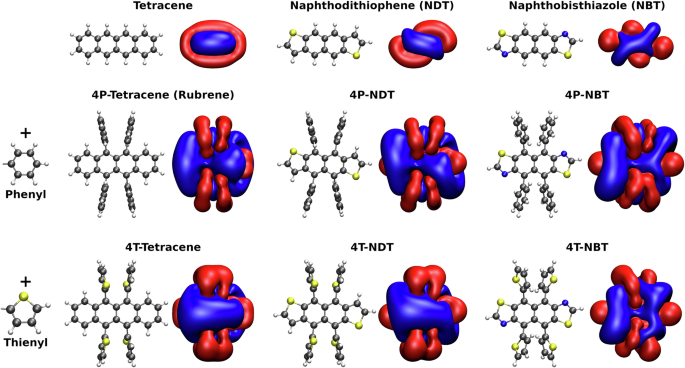
The substitution of heteroatoms and the functionalisation of molecules are established strategies in chemical synthesis. They target the precise tuning of the electronic properties of hydrocarbon molecules to improve their performance in various applications and increase their versatility. Modifications to the molecular structure often lead to simultaneous changes in the morphology such as different crystal structures. These changes can have a stronger and unpredictable impact on the targeted property. The complex relationships between substitution/functionalization in chemical synthesis and the resulting modifications of properties in thin films or crystals are difficult to predict and remain elusive. Here we address these effects for charge carrier transport in organic crystals by combining simulations of carrier mobilities with crystal structure prediction based on density functional theory and density functional tight binding theory. This enables the prediction of carrier mobilities based solely on the molecular structure and allows for the investigation of chemical modifications prior to synthesis and characterisation. Studying nine specific molecules with tetracene and rubrene as reference compounds along with their combined modifications of the molecular cores and additional functionalisations, we unveil systematic trends for the carrier mobilities of their polymorphs. The positive effect of phenyl groups that is responsible for the marked differences between tetracene and rubrene can be transferred to other small molecules such as NDT and NBT leading to a mobility increase by large factors of about five.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
