Manik Garg, Marcin Karpinski, Dorota Matelska, Lawrence Middleton, Oliver S. Burren, Fengyuan Hu, Eleanor Wheeler, Katherine R. Smith, Margarete A. Fabre, Jonathan Mitchell, Amanda O’Neill, Euan A. Ashley, Andrew R. Harper, Quanli Wang, Ryan S. Dhindsa, Slavé Petrovski, Dimitrios Vitsios
{"title":"利用多组学和生物标志物进行疾病预测有助于英国生物库中的病例对照基因发现","authors":"Manik Garg, Marcin Karpinski, Dorota Matelska, Lawrence Middleton, Oliver S. Burren, Fengyuan Hu, Eleanor Wheeler, Katherine R. Smith, Margarete A. Fabre, Jonathan Mitchell, Amanda O’Neill, Euan A. Ashley, Andrew R. Harper, Quanli Wang, Ryan S. Dhindsa, Slavé Petrovski, Dimitrios Vitsios","doi":"10.1038/s41588-024-01898-1","DOIUrl":null,"url":null,"abstract":"The emergence of biobank-level datasets offers new opportunities to discover novel biomarkers and develop predictive algorithms for human disease. Here, we present an ensemble machine-learning framework (machine learning with phenotype associations, MILTON) utilizing a range of biomarkers to predict 3,213 diseases in the UK Biobank. Leveraging the UK Biobank’s longitudinal health record data, MILTON predicts incident disease cases undiagnosed at time of recruitment, largely outperforming available polygenic risk scores. We further demonstrate the utility of MILTON in augmenting genetic association analyses in a phenome-wide association study of 484,230 genome-sequenced samples, along with 46,327 samples with matched plasma proteomics data. This resulted in improved signals for 88 known (P < 1 × 10−8) gene–disease relationships alongside 182 gene–disease relationships that did not achieve genome-wide significance in the nonaugmented baseline cohorts. We validated these discoveries in the FinnGen biobank alongside two orthogonal machine-learning methods built for gene–disease prioritization. All extracted gene–disease associations and incident disease predictive biomarkers are publicly available ( http://milton.public.cgr.astrazeneca.com ). MILTON uses phenotype information in the UK Biobank to identify clinical biomarkers and other quantitative traits that characterize diseases. It then constructs augmented cohorts by predicting undiagnosed individuals, improving power to discover gene–disease relationships.","PeriodicalId":18985,"journal":{"name":"Nature genetics","volume":"56 9","pages":"1821-1831"},"PeriodicalIF":29.0000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41588-024-01898-1.pdf","citationCount":"0","resultStr":"{\"title\":\"Disease prediction with multi-omics and biomarkers empowers case–control genetic discoveries in the UK Biobank\",\"authors\":\"Manik Garg, Marcin Karpinski, Dorota Matelska, Lawrence Middleton, Oliver S. Burren, Fengyuan Hu, Eleanor Wheeler, Katherine R. Smith, Margarete A. Fabre, Jonathan Mitchell, Amanda O’Neill, Euan A. Ashley, Andrew R. Harper, Quanli Wang, Ryan S. Dhindsa, Slavé Petrovski, Dimitrios Vitsios\",\"doi\":\"10.1038/s41588-024-01898-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The emergence of biobank-level datasets offers new opportunities to discover novel biomarkers and develop predictive algorithms for human disease. Here, we present an ensemble machine-learning framework (machine learning with phenotype associations, MILTON) utilizing a range of biomarkers to predict 3,213 diseases in the UK Biobank. Leveraging the UK Biobank’s longitudinal health record data, MILTON predicts incident disease cases undiagnosed at time of recruitment, largely outperforming available polygenic risk scores. We further demonstrate the utility of MILTON in augmenting genetic association analyses in a phenome-wide association study of 484,230 genome-sequenced samples, along with 46,327 samples with matched plasma proteomics data. This resulted in improved signals for 88 known (P < 1 × 10−8) gene–disease relationships alongside 182 gene–disease relationships that did not achieve genome-wide significance in the nonaugmented baseline cohorts. We validated these discoveries in the FinnGen biobank alongside two orthogonal machine-learning methods built for gene–disease prioritization. All extracted gene–disease associations and incident disease predictive biomarkers are publicly available ( http://milton.public.cgr.astrazeneca.com ). MILTON uses phenotype information in the UK Biobank to identify clinical biomarkers and other quantitative traits that characterize diseases. It then constructs augmented cohorts by predicting undiagnosed individuals, improving power to discover gene–disease relationships.\",\"PeriodicalId\":18985,\"journal\":{\"name\":\"Nature genetics\",\"volume\":\"56 9\",\"pages\":\"1821-1831\"},\"PeriodicalIF\":29.0000,\"publicationDate\":\"2024-09-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s41588-024-01898-1.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s41588-024-01898-1\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41588-024-01898-1","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Disease prediction with multi-omics and biomarkers empowers case–control genetic discoveries in the UK Biobank
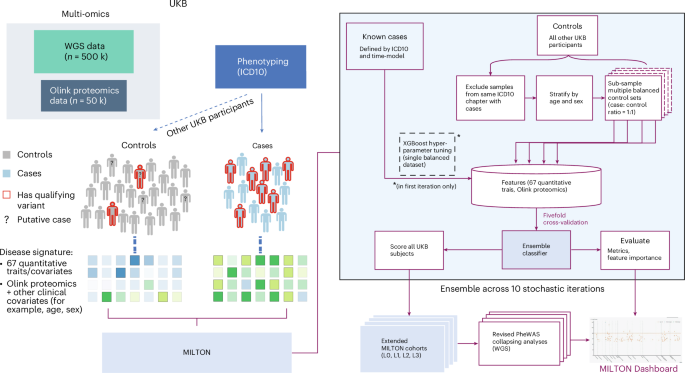
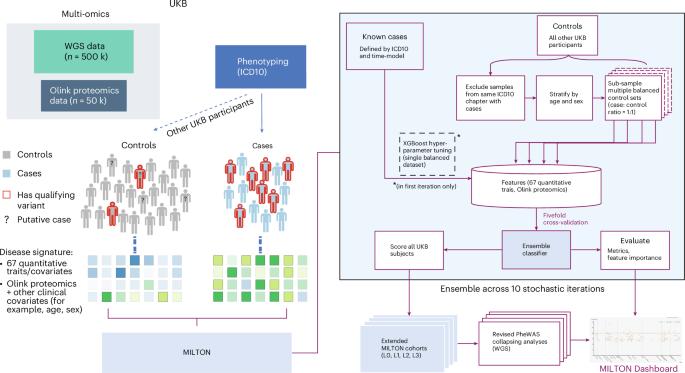
The emergence of biobank-level datasets offers new opportunities to discover novel biomarkers and develop predictive algorithms for human disease. Here, we present an ensemble machine-learning framework (machine learning with phenotype associations, MILTON) utilizing a range of biomarkers to predict 3,213 diseases in the UK Biobank. Leveraging the UK Biobank’s longitudinal health record data, MILTON predicts incident disease cases undiagnosed at time of recruitment, largely outperforming available polygenic risk scores. We further demonstrate the utility of MILTON in augmenting genetic association analyses in a phenome-wide association study of 484,230 genome-sequenced samples, along with 46,327 samples with matched plasma proteomics data. This resulted in improved signals for 88 known (P < 1 × 10−8) gene–disease relationships alongside 182 gene–disease relationships that did not achieve genome-wide significance in the nonaugmented baseline cohorts. We validated these discoveries in the FinnGen biobank alongside two orthogonal machine-learning methods built for gene–disease prioritization. All extracted gene–disease associations and incident disease predictive biomarkers are publicly available ( http://milton.public.cgr.astrazeneca.com ). MILTON uses phenotype information in the UK Biobank to identify clinical biomarkers and other quantitative traits that characterize diseases. It then constructs augmented cohorts by predicting undiagnosed individuals, improving power to discover gene–disease relationships.
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
