Xinyu Peng, Jiaojiao Liang, Kuo Wang, Xiaojie Zhao, Zhiyan Peng, Zhennan Li, Jinhui Zeng, Zheng Lan, Min Lei, Di Huang
{"title":"利用迁移学习为有机材料构建前沿分子轨道预测模型","authors":"Xinyu Peng, Jiaojiao Liang, Kuo Wang, Xiaojie Zhao, Zhiyan Peng, Zhennan Li, Jinhui Zeng, Zheng Lan, Min Lei, Di Huang","doi":"10.1038/s41524-024-01403-6","DOIUrl":null,"url":null,"abstract":"<p>The frontier molecular orbitals of organic semiconductor materials play a crucial role in the performance of photoelectric devices, including organic photovoltaics (OPVs), organic light-emitting diodes (OLEDs), and organic photodetectors (OPDs). In this work, a model for predicting frontier molecular orbital of organic materials, including HOMO and LUMO levels, is established with the extreme gradient boosting algorithm and Klekota-Roth fingerprints. The correlation coefficients of HOMO or LUMO energy levels in the testing set are 0.75 and 0.84 in the transfer model from 11,626 DFT data in Harvard Energy database to 1198 experimental data in literature. The difference between the ML predicted value and the experimental value is smaller than the difference between ML prediction and DFT calculation, always less than 10%. Moreover, based on correlation and SHAP interpretability analysis, 13 key structural fragments influencing energy levels are selected to further verify the effective regulation of the frontier molecular orbital by the key structural fragments in practical applications. Considering the completely opposite regulatory functions of key structural fragments on HOMO and LUMO energy levels, four new Y6 derivatives, Y-PCP, Y-P6F, Y-PCF, and Y-P4FC, are designed to flexibly modify the HOMO and LUMO energy levels. The prediction trends of ML align closely with the computational trends from DFT. It is worth noting that the accuracy of LUMO energy level prediction by the prediction model makes up for the instability of DFT calculation on LUMO energy level. This work offers a cost-effective method to accelerate the acquisition of electronic properties of organic materials.</p>","PeriodicalId":19342,"journal":{"name":"npj Computational Materials","volume":"104 1","pages":""},"PeriodicalIF":11.9000,"publicationDate":"2024-09-11","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Construction frontier molecular orbital prediction model with transfer learning for organic materials\",\"authors\":\"Xinyu Peng, Jiaojiao Liang, Kuo Wang, Xiaojie Zhao, Zhiyan Peng, Zhennan Li, Jinhui Zeng, Zheng Lan, Min Lei, Di Huang\",\"doi\":\"10.1038/s41524-024-01403-6\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The frontier molecular orbitals of organic semiconductor materials play a crucial role in the performance of photoelectric devices, including organic photovoltaics (OPVs), organic light-emitting diodes (OLEDs), and organic photodetectors (OPDs). In this work, a model for predicting frontier molecular orbital of organic materials, including HOMO and LUMO levels, is established with the extreme gradient boosting algorithm and Klekota-Roth fingerprints. The correlation coefficients of HOMO or LUMO energy levels in the testing set are 0.75 and 0.84 in the transfer model from 11,626 DFT data in Harvard Energy database to 1198 experimental data in literature. The difference between the ML predicted value and the experimental value is smaller than the difference between ML prediction and DFT calculation, always less than 10%. Moreover, based on correlation and SHAP interpretability analysis, 13 key structural fragments influencing energy levels are selected to further verify the effective regulation of the frontier molecular orbital by the key structural fragments in practical applications. Considering the completely opposite regulatory functions of key structural fragments on HOMO and LUMO energy levels, four new Y6 derivatives, Y-PCP, Y-P6F, Y-PCF, and Y-P4FC, are designed to flexibly modify the HOMO and LUMO energy levels. The prediction trends of ML align closely with the computational trends from DFT. It is worth noting that the accuracy of LUMO energy level prediction by the prediction model makes up for the instability of DFT calculation on LUMO energy level. This work offers a cost-effective method to accelerate the acquisition of electronic properties of organic materials.</p>\",\"PeriodicalId\":19342,\"journal\":{\"name\":\"npj Computational Materials\",\"volume\":\"104 1\",\"pages\":\"\"},\"PeriodicalIF\":11.9000,\"publicationDate\":\"2024-09-11\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"npj Computational Materials\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://doi.org/10.1038/s41524-024-01403-6\",\"RegionNum\":1,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"npj Computational Materials","FirstCategoryId":"88","ListUrlMain":"https://doi.org/10.1038/s41524-024-01403-6","RegionNum":1,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Construction frontier molecular orbital prediction model with transfer learning for organic materials
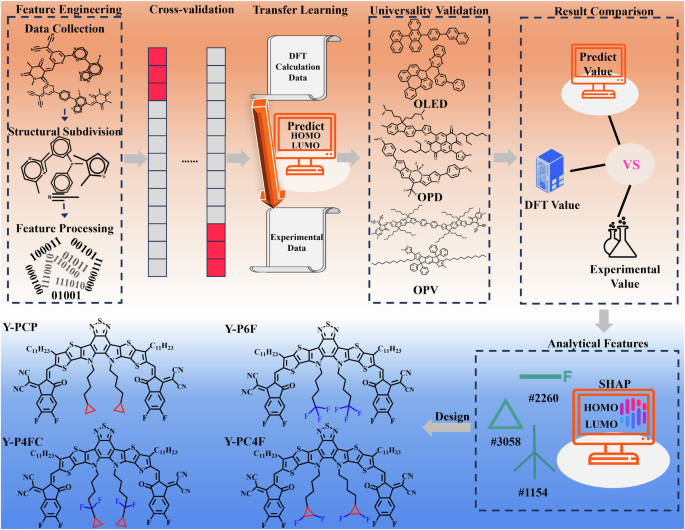
The frontier molecular orbitals of organic semiconductor materials play a crucial role in the performance of photoelectric devices, including organic photovoltaics (OPVs), organic light-emitting diodes (OLEDs), and organic photodetectors (OPDs). In this work, a model for predicting frontier molecular orbital of organic materials, including HOMO and LUMO levels, is established with the extreme gradient boosting algorithm and Klekota-Roth fingerprints. The correlation coefficients of HOMO or LUMO energy levels in the testing set are 0.75 and 0.84 in the transfer model from 11,626 DFT data in Harvard Energy database to 1198 experimental data in literature. The difference between the ML predicted value and the experimental value is smaller than the difference between ML prediction and DFT calculation, always less than 10%. Moreover, based on correlation and SHAP interpretability analysis, 13 key structural fragments influencing energy levels are selected to further verify the effective regulation of the frontier molecular orbital by the key structural fragments in practical applications. Considering the completely opposite regulatory functions of key structural fragments on HOMO and LUMO energy levels, four new Y6 derivatives, Y-PCP, Y-P6F, Y-PCF, and Y-P4FC, are designed to flexibly modify the HOMO and LUMO energy levels. The prediction trends of ML align closely with the computational trends from DFT. It is worth noting that the accuracy of LUMO energy level prediction by the prediction model makes up for the instability of DFT calculation on LUMO energy level. This work offers a cost-effective method to accelerate the acquisition of electronic properties of organic materials.
期刊介绍:
npj Computational Materials is a high-quality open access journal from Nature Research that publishes research papers applying computational approaches for the design of new materials and enhancing our understanding of existing ones. The journal also welcomes papers on new computational techniques and the refinement of current approaches that support these aims, as well as experimental papers that complement computational findings.
Some key features of npj Computational Materials include a 2-year impact factor of 12.241 (2021), article downloads of 1,138,590 (2021), and a fast turnaround time of 11 days from submission to the first editorial decision. The journal is indexed in various databases and services, including Chemical Abstracts Service (ACS), Astrophysics Data System (ADS), Current Contents/Physical, Chemical and Earth Sciences, Journal Citation Reports/Science Edition, SCOPUS, EI Compendex, INSPEC, Google Scholar, SCImago, DOAJ, CNKI, and Science Citation Index Expanded (SCIE), among others.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
