Maryam Abdinejad, Amirhossein Farzi, Robin Möller-Gulland, Fokko Mulder, Chengyu Liu, Junming Shao, Jasper Biemolt, Marc Robert, Ali Seifitokaldani, Thomas Burdyny
{"title":"消除支撑分子催化剂上氧化还原介导的电子传递机制,实现二氧化碳到乙醇的转化","authors":"Maryam Abdinejad, Amirhossein Farzi, Robin Möller-Gulland, Fokko Mulder, Chengyu Liu, Junming Shao, Jasper Biemolt, Marc Robert, Ali Seifitokaldani, Thomas Burdyny","doi":"10.1038/s41929-024-01225-1","DOIUrl":null,"url":null,"abstract":"Molecular catalysts play a significant role in chemical transformations, utilizing changes in redox states to facilitate reactions. To date molecular electrocatalysts have efficiently produced single-carbon products from CO2 but have struggled to achieve a carbon–carbon coupling step. Conversely, copper catalysts can enable carbon–carbon coupling, but lead to broad C2+ product spectra. Here we subvert the traditional redox-mediated reaction mechanisms of organometallic compounds through a heterogeneous nickel-supported iron tetraphenylporphyrin electrocatalyst, facilitating electrochemical carbon–carbon coupling to produce ethanol. This represents a marked behavioural shift compared with carbon-supported metalloporphyrins. Extending the approach to a three-dimensional porous nickel support with adsorbed iron tetraphenylporphyrin, we attain ethanol Faradaic efficiencies of 68% ± 3.2% at −0.3 V versus a reversible hydrogen electrode (pH 7.7) with partial ethanol current densities of −21 mA cm−2. Separately we demonstrate maintained ethanol production over 60 h of operation. Further consideration of the wide parameter space of molecular catalyst and metal electrodes shows promise for additional chemistries and achievable metrics. The electrochemical reduction of CO2 on organometallic catalysts is commonly limited to two-electron products. Now, an iron tetraphenylporphyrin catalyst immobilized onto a nickel electrode is shown to achieve a Faradaic efficiency for ethanol of 68% due to the strong electronic coupling between the catalyst and the support.","PeriodicalId":18845,"journal":{"name":"Nature Catalysis","volume":"7 10","pages":"1109-1119"},"PeriodicalIF":44.6000,"publicationDate":"2024-09-13","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Eliminating redox-mediated electron transfer mechanisms on a supported molecular catalyst enables CO2 conversion to ethanol\",\"authors\":\"Maryam Abdinejad, Amirhossein Farzi, Robin Möller-Gulland, Fokko Mulder, Chengyu Liu, Junming Shao, Jasper Biemolt, Marc Robert, Ali Seifitokaldani, Thomas Burdyny\",\"doi\":\"10.1038/s41929-024-01225-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Molecular catalysts play a significant role in chemical transformations, utilizing changes in redox states to facilitate reactions. To date molecular electrocatalysts have efficiently produced single-carbon products from CO2 but have struggled to achieve a carbon–carbon coupling step. Conversely, copper catalysts can enable carbon–carbon coupling, but lead to broad C2+ product spectra. Here we subvert the traditional redox-mediated reaction mechanisms of organometallic compounds through a heterogeneous nickel-supported iron tetraphenylporphyrin electrocatalyst, facilitating electrochemical carbon–carbon coupling to produce ethanol. This represents a marked behavioural shift compared with carbon-supported metalloporphyrins. Extending the approach to a three-dimensional porous nickel support with adsorbed iron tetraphenylporphyrin, we attain ethanol Faradaic efficiencies of 68% ± 3.2% at −0.3 V versus a reversible hydrogen electrode (pH 7.7) with partial ethanol current densities of −21 mA cm−2. Separately we demonstrate maintained ethanol production over 60 h of operation. Further consideration of the wide parameter space of molecular catalyst and metal electrodes shows promise for additional chemistries and achievable metrics. The electrochemical reduction of CO2 on organometallic catalysts is commonly limited to two-electron products. Now, an iron tetraphenylporphyrin catalyst immobilized onto a nickel electrode is shown to achieve a Faradaic efficiency for ethanol of 68% due to the strong electronic coupling between the catalyst and the support.\",\"PeriodicalId\":18845,\"journal\":{\"name\":\"Nature Catalysis\",\"volume\":\"7 10\",\"pages\":\"1109-1119\"},\"PeriodicalIF\":44.6000,\"publicationDate\":\"2024-09-13\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature Catalysis\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.nature.com/articles/s41929-024-01225-1\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature Catalysis","FirstCategoryId":"92","ListUrlMain":"https://www.nature.com/articles/s41929-024-01225-1","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Eliminating redox-mediated electron transfer mechanisms on a supported molecular catalyst enables CO2 conversion to ethanol
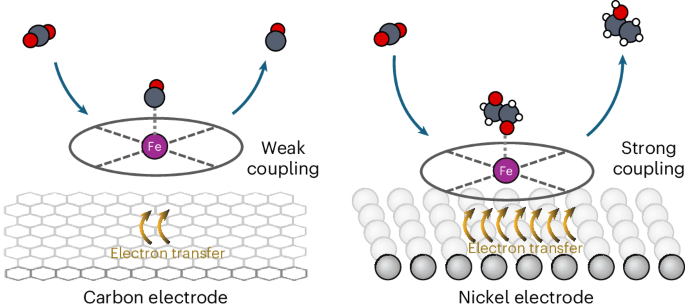
Molecular catalysts play a significant role in chemical transformations, utilizing changes in redox states to facilitate reactions. To date molecular electrocatalysts have efficiently produced single-carbon products from CO2 but have struggled to achieve a carbon–carbon coupling step. Conversely, copper catalysts can enable carbon–carbon coupling, but lead to broad C2+ product spectra. Here we subvert the traditional redox-mediated reaction mechanisms of organometallic compounds through a heterogeneous nickel-supported iron tetraphenylporphyrin electrocatalyst, facilitating electrochemical carbon–carbon coupling to produce ethanol. This represents a marked behavioural shift compared with carbon-supported metalloporphyrins. Extending the approach to a three-dimensional porous nickel support with adsorbed iron tetraphenylporphyrin, we attain ethanol Faradaic efficiencies of 68% ± 3.2% at −0.3 V versus a reversible hydrogen electrode (pH 7.7) with partial ethanol current densities of −21 mA cm−2. Separately we demonstrate maintained ethanol production over 60 h of operation. Further consideration of the wide parameter space of molecular catalyst and metal electrodes shows promise for additional chemistries and achievable metrics. The electrochemical reduction of CO2 on organometallic catalysts is commonly limited to two-electron products. Now, an iron tetraphenylporphyrin catalyst immobilized onto a nickel electrode is shown to achieve a Faradaic efficiency for ethanol of 68% due to the strong electronic coupling between the catalyst and the support.
期刊介绍:
Nature Catalysis serves as a platform for researchers across chemistry and related fields, focusing on homogeneous catalysis, heterogeneous catalysis, and biocatalysts, encompassing both fundamental and applied studies. With a particular emphasis on advancing sustainable industries and processes, the journal provides comprehensive coverage of catalysis research, appealing to scientists, engineers, and researchers in academia and industry.
Maintaining the high standards of the Nature brand, Nature Catalysis boasts a dedicated team of professional editors, rigorous peer-review processes, and swift publication times, ensuring editorial independence and quality. The journal publishes work spanning heterogeneous catalysis, homogeneous catalysis, and biocatalysis, covering areas such as catalytic synthesis, mechanisms, characterization, computational studies, nanoparticle catalysis, electrocatalysis, photocatalysis, environmental catalysis, asymmetric catalysis, and various forms of organocatalysis.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
