{"title":"从第一原理计算看 Ag/SnO2 界面的粘附性、稳定性和电子特性","authors":"Yunhui Xu, Jintao Li, Wensong Teng, Defeng Cui, Xiaolong Zhou","doi":"10.1002/crat.202400126","DOIUrl":null,"url":null,"abstract":"<p>The interfacial bonding state between each oxide and the silver matrix in AgCuOIn<sub>2</sub>O<sub>3</sub>SnO<sub>2</sub> electrical contact materials remains unclear. To address this, first-principles calculations using density-functional theory are employed to establish the low-index surfaces of Ag and SnO<sub>2</sub> and perform convergence tests. Computational results reveal that the Ag (111) surface and the SnO<sub>2</sub>(110)-O surface exhibit the highest stability among their respective low-index surfaces. Consequently, these surfaces are chosen to form the interfacial model, and their atomic structure, adhesion work, and interfacial energies are systematically analyzed. The results demonstrate that the stability and interfacial bonding strength of the Ag(111)/SnO<sub>2</sub>(110)-O interface are high, exhibiting metallic properties and strong conductivity. Moreover, at an interface spacing of d<sub>0</sub> = 2.4 Å, the interface stability is optimal. The redistribution of charge at the interface induces significant changes in the local atomic density of states, particularly noticeable in the Ag and O atoms. Additionally, the Ag/SnO<sub>2</sub> interface is predominantly bonded through ionic interactions, contributing to its robust bonding.</p>","PeriodicalId":48935,"journal":{"name":"Crystal Research and Technology","volume":"59 11","pages":""},"PeriodicalIF":1.9000,"publicationDate":"2024-08-21","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Adhesion, Stability and Electronic Properties of Ag/SnO2 Interface from First-Principles Calculation\",\"authors\":\"Yunhui Xu, Jintao Li, Wensong Teng, Defeng Cui, Xiaolong Zhou\",\"doi\":\"10.1002/crat.202400126\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The interfacial bonding state between each oxide and the silver matrix in AgCuOIn<sub>2</sub>O<sub>3</sub>SnO<sub>2</sub> electrical contact materials remains unclear. To address this, first-principles calculations using density-functional theory are employed to establish the low-index surfaces of Ag and SnO<sub>2</sub> and perform convergence tests. Computational results reveal that the Ag (111) surface and the SnO<sub>2</sub>(110)-O surface exhibit the highest stability among their respective low-index surfaces. Consequently, these surfaces are chosen to form the interfacial model, and their atomic structure, adhesion work, and interfacial energies are systematically analyzed. The results demonstrate that the stability and interfacial bonding strength of the Ag(111)/SnO<sub>2</sub>(110)-O interface are high, exhibiting metallic properties and strong conductivity. Moreover, at an interface spacing of d<sub>0</sub> = 2.4 Å, the interface stability is optimal. The redistribution of charge at the interface induces significant changes in the local atomic density of states, particularly noticeable in the Ag and O atoms. Additionally, the Ag/SnO<sub>2</sub> interface is predominantly bonded through ionic interactions, contributing to its robust bonding.</p>\",\"PeriodicalId\":48935,\"journal\":{\"name\":\"Crystal Research and Technology\",\"volume\":\"59 11\",\"pages\":\"\"},\"PeriodicalIF\":1.9000,\"publicationDate\":\"2024-08-21\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Crystal Research and Technology\",\"FirstCategoryId\":\"88\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/crat.202400126\",\"RegionNum\":4,\"RegionCategory\":\"材料科学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"Chemistry\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Crystal Research and Technology","FirstCategoryId":"88","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/crat.202400126","RegionNum":4,"RegionCategory":"材料科学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"Chemistry","Score":null,"Total":0}
引用次数: 0
摘要
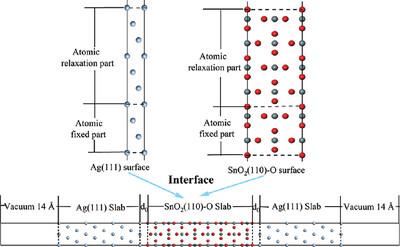
AgCuOIn2O3SnO2 电接触材料中每种氧化物与银基体之间的界面结合状态仍不清楚。为了解决这个问题,我们采用密度泛函理论进行了第一原理计算,建立了银和二氧化锡的低指数表面,并进行了收敛性测试。计算结果表明,在各自的低指数表面中,Ag (111) 表面和 SnO2(110)-O 表面表现出最高的稳定性。因此,我们选择这些表面组成界面模型,并系统分析了它们的原子结构、粘附功和界面能。结果表明,Ag(111)/SnO2(110)-O 界面具有很高的稳定性和界面结合强度,表现出金属特性和很强的导电性。此外,当界面间距为 d0 = 2.4 Å 时,界面稳定性最佳。界面上的电荷再分布引起了局部原子态密度的显著变化,这在 Ag 原子和 O 原子中尤为明显。此外,Ag/SnO2 界面主要通过离子相互作用成键,这也是其稳健成键的原因之一。
Adhesion, Stability and Electronic Properties of Ag/SnO2 Interface from First-Principles Calculation
The interfacial bonding state between each oxide and the silver matrix in AgCuOIn2O3SnO2 electrical contact materials remains unclear. To address this, first-principles calculations using density-functional theory are employed to establish the low-index surfaces of Ag and SnO2 and perform convergence tests. Computational results reveal that the Ag (111) surface and the SnO2(110)-O surface exhibit the highest stability among their respective low-index surfaces. Consequently, these surfaces are chosen to form the interfacial model, and their atomic structure, adhesion work, and interfacial energies are systematically analyzed. The results demonstrate that the stability and interfacial bonding strength of the Ag(111)/SnO2(110)-O interface are high, exhibiting metallic properties and strong conductivity. Moreover, at an interface spacing of d0 = 2.4 Å, the interface stability is optimal. The redistribution of charge at the interface induces significant changes in the local atomic density of states, particularly noticeable in the Ag and O atoms. Additionally, the Ag/SnO2 interface is predominantly bonded through ionic interactions, contributing to its robust bonding.
期刊介绍:
The journal Crystal Research and Technology is a pure online Journal (since 2012).
Crystal Research and Technology is an international journal examining all aspects of research within experimental, industrial, and theoretical crystallography. The journal covers the relevant aspects of
-crystal growth techniques and phenomena (including bulk growth, thin films)
-modern crystalline materials (e.g. smart materials, nanocrystals, quasicrystals, liquid crystals)
-industrial crystallisation
-application of crystals in materials science, electronics, data storage, and optics
-experimental, simulation and theoretical studies of the structural properties of crystals
-crystallographic computing



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
