Michelle T. Custodio Castro, Carlos O. Della Védova, Rosana M. Romano
{"title":"探索 XC(W)ZY 分子中的构象偏好,其中 X、Y = F、Cl、Br 和 W、Z = O、S、Se:揭示共轭和同分异构相互作用的影响","authors":"Michelle T. Custodio Castro, Carlos O. Della Védova, Rosana M. Romano","doi":"10.1002/poc.4654","DOIUrl":null,"url":null,"abstract":"<div>\n \n <p>The relative stabilities of the <i>syn</i>- and <i>anti</i>-conformers of 72 molecules belonging to the XC(W)ZY type, with X, Y = F, Cl, Br and W, Z = O, S, Se, have been computed using the B3LYP/aug-cc-pVDZ approximation. The conformational preferences, represented by the energy differences between the two rotamers, exhibit a systematic trend in relation to both the halogen atoms and the chalcogen atoms. These computational predictions are in agreement with available experimental results. The NBO formalism was employed to assess the influence of both the conjugative and anomeric interactions on the relative energy of the conformers. It has been determined that the conjugative interaction provides a satisfactory explanation for the energy differences between rotamers. In contrast, the anomeric interactions favors the <i>syn</i>-conformation in all cases. The relative stabilities between XC(W)ZY/YC(W)ZX and XC(W)ZY/XC(Z)WY constitutional isomers have also been computed and correlated with the experimental data.</p>\n </div>","PeriodicalId":16829,"journal":{"name":"Journal of Physical Organic Chemistry","volume":"37 11","pages":""},"PeriodicalIF":1.8000,"publicationDate":"2024-08-12","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring Conformational Preferences in XC(W)ZY Molecules With X, Y = F, Cl, Br and W, Z = O, S, Se: Unraveling the Influence of Conjugative and Anomeric Interactions\",\"authors\":\"Michelle T. Custodio Castro, Carlos O. Della Védova, Rosana M. Romano\",\"doi\":\"10.1002/poc.4654\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div>\\n \\n <p>The relative stabilities of the <i>syn</i>- and <i>anti</i>-conformers of 72 molecules belonging to the XC(W)ZY type, with X, Y = F, Cl, Br and W, Z = O, S, Se, have been computed using the B3LYP/aug-cc-pVDZ approximation. The conformational preferences, represented by the energy differences between the two rotamers, exhibit a systematic trend in relation to both the halogen atoms and the chalcogen atoms. These computational predictions are in agreement with available experimental results. The NBO formalism was employed to assess the influence of both the conjugative and anomeric interactions on the relative energy of the conformers. It has been determined that the conjugative interaction provides a satisfactory explanation for the energy differences between rotamers. In contrast, the anomeric interactions favors the <i>syn</i>-conformation in all cases. The relative stabilities between XC(W)ZY/YC(W)ZX and XC(W)ZY/XC(Z)WY constitutional isomers have also been computed and correlated with the experimental data.</p>\\n </div>\",\"PeriodicalId\":16829,\"journal\":{\"name\":\"Journal of Physical Organic Chemistry\",\"volume\":\"37 11\",\"pages\":\"\"},\"PeriodicalIF\":1.8000,\"publicationDate\":\"2024-08-12\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Physical Organic Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/poc.4654\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, ORGANIC\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Physical Organic Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/poc.4654","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, ORGANIC","Score":null,"Total":0}
Exploring Conformational Preferences in XC(W)ZY Molecules With X, Y = F, Cl, Br and W, Z = O, S, Se: Unraveling the Influence of Conjugative and Anomeric Interactions
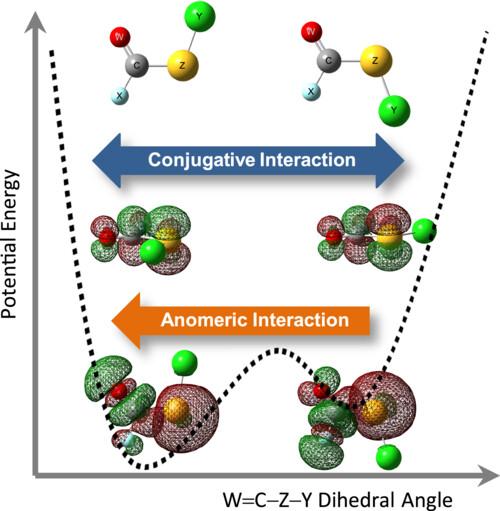
The relative stabilities of the syn- and anti-conformers of 72 molecules belonging to the XC(W)ZY type, with X, Y = F, Cl, Br and W, Z = O, S, Se, have been computed using the B3LYP/aug-cc-pVDZ approximation. The conformational preferences, represented by the energy differences between the two rotamers, exhibit a systematic trend in relation to both the halogen atoms and the chalcogen atoms. These computational predictions are in agreement with available experimental results. The NBO formalism was employed to assess the influence of both the conjugative and anomeric interactions on the relative energy of the conformers. It has been determined that the conjugative interaction provides a satisfactory explanation for the energy differences between rotamers. In contrast, the anomeric interactions favors the syn-conformation in all cases. The relative stabilities between XC(W)ZY/YC(W)ZX and XC(W)ZY/XC(Z)WY constitutional isomers have also been computed and correlated with the experimental data.
期刊介绍:
The Journal of Physical Organic Chemistry is the foremost international journal devoted to the relationship between molecular structure and chemical reactivity in organic systems. It publishes Research Articles, Reviews and Mini Reviews based on research striving to understand the principles governing chemical structures in relation to activity and transformation with physical and mathematical rigor, using results derived from experimental and computational methods. Physical Organic Chemistry is a central and fundamental field with multiple applications in fields such as molecular recognition, supramolecular chemistry, catalysis, photochemistry, biological and material sciences, nanotechnology and surface science.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
