Arpita Deb , Brian D. Tow , Jie Hao , Branden L. Nguyen , Valeria Gomez , James A. Stewart Jr , Ashley J. Smuder , Bjorn C. Knollmann , Ying Wang , Bin Liu
{"title":"在 CPVT 的遗传性心律失常模型中,MCU 的条件性消融会加重心脏病理变化","authors":"Arpita Deb , Brian D. Tow , Jie Hao , Branden L. Nguyen , Valeria Gomez , James A. Stewart Jr , Ashley J. Smuder , Bjorn C. Knollmann , Ying Wang , Bin Liu","doi":"10.1016/j.jmccpl.2024.100093","DOIUrl":null,"url":null,"abstract":"<div><h3>Background</h3><p>Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetic arrhythmic syndrome caused by mutations in the calcium (Ca<sup>2+</sup>) release channel ryanodine receptor (RyR2) and its accessory proteins. These mutations make the channel leaky, resulting in Ca<sup>2+</sup>-dependent arrhythmias. Besides arrhythmias, CPVT hearts typically lack structural cardiac remodeling, a characteristic often observed in other cardiac conditions (heart failure, prediabetes) also marked by RyR2 leak. Recent studies suggest that mitochondria are able to accommodate more Ca<sup>2+</sup> influx to inhibit arrhythmias in CPVT. Thus, we hypothesize that CPVT mitochondria can absorb diastolic Ca<sup>2+</sup> to protect the heart from cardiac remodeling.</p></div><div><h3>Methods and results</h3><p>The Mitochondrial Ca<sup>2+</sup> uniporter (MCU), the main mitochondrial Ca<sup>2+</sup> uptake protein, was conditionally knocked out in a CPVT model of calsequestrin 2 (CASQ2) KO. In vivo cardiac function was impaired in the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> model as assessed by echocardiography. Cardiac dilation and cellular hypertrophy were also observed in the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> hearts. Live-cell imaging identified altered Ca<sup>2+</sup> handling and increased oxidative stress in CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> myocytes. The activation status of Ca<sup>2+</sup>-dependent remodeling pathways (CaMKII, Calcineurin) was not altered in the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> model. RNAseq identified changes in the transcriptome of the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> hearts, distinct from the classic cardiac remodeling program of fetal gene re-expression.</p></div><div><h3>Conclusions</h3><p>We present genetic evidence that mitochondria play a protective role in CPVT. MCU-dependent Ca<sup>2+</sup> uptake is crucial for preventing pathological cardiac remodeling in CPVT.</p></div>","PeriodicalId":73835,"journal":{"name":"Journal of molecular and cellular cardiology plus","volume":"10 ","pages":"Article 100093"},"PeriodicalIF":2.2000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.sciencedirect.com/science/article/pii/S2772976124000333/pdfft?md5=8ef6c8a61076f2dcb4155d45ed6349eb&pid=1-s2.0-S2772976124000333-main.pdf","citationCount":"0","resultStr":"{\"title\":\"Conditional ablation of MCU exacerbated cardiac pathology in a genetic arrhythmic model of CPVT\",\"authors\":\"Arpita Deb , Brian D. Tow , Jie Hao , Branden L. Nguyen , Valeria Gomez , James A. Stewart Jr , Ashley J. Smuder , Bjorn C. Knollmann , Ying Wang , Bin Liu\",\"doi\":\"10.1016/j.jmccpl.2024.100093\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background</h3><p>Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetic arrhythmic syndrome caused by mutations in the calcium (Ca<sup>2+</sup>) release channel ryanodine receptor (RyR2) and its accessory proteins. These mutations make the channel leaky, resulting in Ca<sup>2+</sup>-dependent arrhythmias. Besides arrhythmias, CPVT hearts typically lack structural cardiac remodeling, a characteristic often observed in other cardiac conditions (heart failure, prediabetes) also marked by RyR2 leak. Recent studies suggest that mitochondria are able to accommodate more Ca<sup>2+</sup> influx to inhibit arrhythmias in CPVT. Thus, we hypothesize that CPVT mitochondria can absorb diastolic Ca<sup>2+</sup> to protect the heart from cardiac remodeling.</p></div><div><h3>Methods and results</h3><p>The Mitochondrial Ca<sup>2+</sup> uniporter (MCU), the main mitochondrial Ca<sup>2+</sup> uptake protein, was conditionally knocked out in a CPVT model of calsequestrin 2 (CASQ2) KO. In vivo cardiac function was impaired in the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> model as assessed by echocardiography. Cardiac dilation and cellular hypertrophy were also observed in the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> hearts. Live-cell imaging identified altered Ca<sup>2+</sup> handling and increased oxidative stress in CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> myocytes. The activation status of Ca<sup>2+</sup>-dependent remodeling pathways (CaMKII, Calcineurin) was not altered in the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> model. RNAseq identified changes in the transcriptome of the CASQ2<sup>−/−</sup>-MCU<sup>CKO</sup> hearts, distinct from the classic cardiac remodeling program of fetal gene re-expression.</p></div><div><h3>Conclusions</h3><p>We present genetic evidence that mitochondria play a protective role in CPVT. MCU-dependent Ca<sup>2+</sup> uptake is crucial for preventing pathological cardiac remodeling in CPVT.</p></div>\",\"PeriodicalId\":73835,\"journal\":{\"name\":\"Journal of molecular and cellular cardiology plus\",\"volume\":\"10 \",\"pages\":\"Article 100093\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.sciencedirect.com/science/article/pii/S2772976124000333/pdfft?md5=8ef6c8a61076f2dcb4155d45ed6349eb&pid=1-s2.0-S2772976124000333-main.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of molecular and cellular cardiology plus\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2772976124000333\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/10 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of molecular and cellular cardiology plus","FirstCategoryId":"1085","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2772976124000333","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/10 0:00:00","PubModel":"Epub","JCR":"","JCRName":"","Score":null,"Total":0}
Conditional ablation of MCU exacerbated cardiac pathology in a genetic arrhythmic model of CPVT
Background
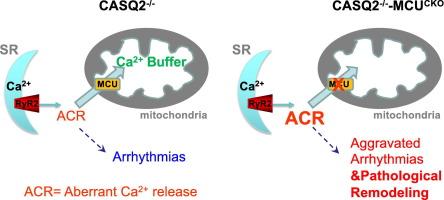
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a genetic arrhythmic syndrome caused by mutations in the calcium (Ca2+) release channel ryanodine receptor (RyR2) and its accessory proteins. These mutations make the channel leaky, resulting in Ca2+-dependent arrhythmias. Besides arrhythmias, CPVT hearts typically lack structural cardiac remodeling, a characteristic often observed in other cardiac conditions (heart failure, prediabetes) also marked by RyR2 leak. Recent studies suggest that mitochondria are able to accommodate more Ca2+ influx to inhibit arrhythmias in CPVT. Thus, we hypothesize that CPVT mitochondria can absorb diastolic Ca2+ to protect the heart from cardiac remodeling.
Methods and results
The Mitochondrial Ca2+ uniporter (MCU), the main mitochondrial Ca2+ uptake protein, was conditionally knocked out in a CPVT model of calsequestrin 2 (CASQ2) KO. In vivo cardiac function was impaired in the CASQ2−/−-MCUCKO model as assessed by echocardiography. Cardiac dilation and cellular hypertrophy were also observed in the CASQ2−/−-MCUCKO hearts. Live-cell imaging identified altered Ca2+ handling and increased oxidative stress in CASQ2−/−-MCUCKO myocytes. The activation status of Ca2+-dependent remodeling pathways (CaMKII, Calcineurin) was not altered in the CASQ2−/−-MCUCKO model. RNAseq identified changes in the transcriptome of the CASQ2−/−-MCUCKO hearts, distinct from the classic cardiac remodeling program of fetal gene re-expression.
Conclusions
We present genetic evidence that mitochondria play a protective role in CPVT. MCU-dependent Ca2+ uptake is crucial for preventing pathological cardiac remodeling in CPVT.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
