{"title":"用高斯和增强平面波方法计算时变密度泛函理论的激发态作用力","authors":"Beliz Sertcan Gökmen, Jürg Hutter, Anna-Sophia Hehn","doi":"10.1021/acs.jctc.4c00614","DOIUrl":null,"url":null,"abstract":"Augmented plane wave methods enable an efficient description of atom-centered or localized features of the electronic density, circumventing high energy cutoffs and thus prohibitive computational costs of pure plane wave formulations. To complement existing implementations for ground-state properties and excitation energies, we present the extension of the Gaussian and augmented plane wave method to excited-state nuclear gradients within the Tamm–Dancoff approximation of time-dependent density functional theory and its implementation in the CP2K program package. Benchmarks for a test set of 35 small molecules demonstrate that maximum errors in the nuclear forces for excited states of singlet and triplet spin multiplicity are smaller than 0.1 eV/Å. The method is furthermore applied to the calculation of the zero-phonon line of defective hexagonal boron nitride. This spectral feature is reproduced with an error of 0.6 eV in comparison to GW–Bethe–Salpeter reference computations and 0.4 eV in comparison to experimental measurements. Accuracy assessments and applications thus demonstrate the potential use of the outlined developments for large-scale applications on excited-state properties of extended systems.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"45 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Excited-State Forces with the Gaussian and Augmented Plane Wave Method for the Tamm–Dancoff Approximation of Time-Dependent Density Functional Theory\",\"authors\":\"Beliz Sertcan Gökmen, Jürg Hutter, Anna-Sophia Hehn\",\"doi\":\"10.1021/acs.jctc.4c00614\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Augmented plane wave methods enable an efficient description of atom-centered or localized features of the electronic density, circumventing high energy cutoffs and thus prohibitive computational costs of pure plane wave formulations. To complement existing implementations for ground-state properties and excitation energies, we present the extension of the Gaussian and augmented plane wave method to excited-state nuclear gradients within the Tamm–Dancoff approximation of time-dependent density functional theory and its implementation in the CP2K program package. Benchmarks for a test set of 35 small molecules demonstrate that maximum errors in the nuclear forces for excited states of singlet and triplet spin multiplicity are smaller than 0.1 eV/Å. The method is furthermore applied to the calculation of the zero-phonon line of defective hexagonal boron nitride. This spectral feature is reproduced with an error of 0.6 eV in comparison to GW–Bethe–Salpeter reference computations and 0.4 eV in comparison to experimental measurements. Accuracy assessments and applications thus demonstrate the potential use of the outlined developments for large-scale applications on excited-state properties of extended systems.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"45 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00614\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00614","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Excited-State Forces with the Gaussian and Augmented Plane Wave Method for the Tamm–Dancoff Approximation of Time-Dependent Density Functional Theory
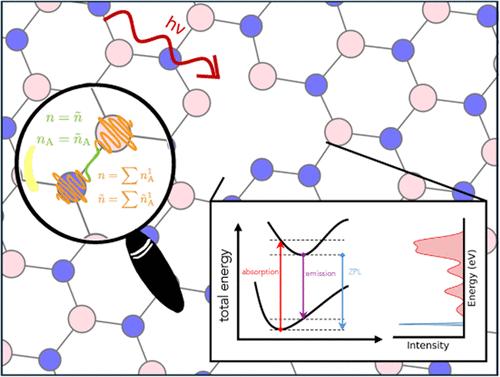
Augmented plane wave methods enable an efficient description of atom-centered or localized features of the electronic density, circumventing high energy cutoffs and thus prohibitive computational costs of pure plane wave formulations. To complement existing implementations for ground-state properties and excitation energies, we present the extension of the Gaussian and augmented plane wave method to excited-state nuclear gradients within the Tamm–Dancoff approximation of time-dependent density functional theory and its implementation in the CP2K program package. Benchmarks for a test set of 35 small molecules demonstrate that maximum errors in the nuclear forces for excited states of singlet and triplet spin multiplicity are smaller than 0.1 eV/Å. The method is furthermore applied to the calculation of the zero-phonon line of defective hexagonal boron nitride. This spectral feature is reproduced with an error of 0.6 eV in comparison to GW–Bethe–Salpeter reference computations and 0.4 eV in comparison to experimental measurements. Accuracy assessments and applications thus demonstrate the potential use of the outlined developments for large-scale applications on excited-state properties of extended systems.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
