{"title":"Massive Assessment of the Geometries of Atmospheric Molecular Clusters","authors":"Andreas Buchgraitz Jensen, Jonas Elm","doi":"10.1021/acs.jctc.4c01046","DOIUrl":null,"url":null,"abstract":"Atmospheric molecular clusters are important for the formation of new aerosol particles in the air. However, current experimental techniques are not able to yield direct insight into the cluster geometries. This implies that to date there is limited information about how accurately the applied computational methods depict the cluster structures. Here we massively benchmark the molecular geometries of atmospheric molecular clusters. We initially assessed how well different DF-MP2 approaches reproduce the geometries of 45 dimer clusters obtained at a high DF-CCSD(T)-F12b/cc-pVDZ-F12 level of theory. Based on the results, we find that the DF-MP2/aug-cc-pVQZ level of theory best resembles the DF-CCSD(T)-F12b/cc-pVDZ-F12 reference level. We subsequently optimized 1283 acid–base cluster structures (up to tetramers) at the DF-MP2/aug-cc-pVQZ level of theory and assessed how more approximate methods reproduce the geometries. Out of the tested semiempirical methods, we find that the newly parametrized atmospheric molecular cluster extended tight binding method (AMC-xTB) is most reliable for locating the correct lowest energy configuration and yields the lowest root mean square deviation (RMSD) compared to the reference level. In addition, we find that the DFT-3c methods show similar performance as the usually employed ωB97X-D/6-31++G(d,p) level of theory at a potentially reduced computational cost. This suggests that these methods could prove to be valuable for large-scale screening of cluster structures in the future.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"31 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":null,"platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01046","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
Abstract
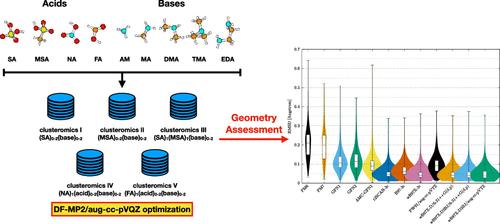
Atmospheric molecular clusters are important for the formation of new aerosol particles in the air. However, current experimental techniques are not able to yield direct insight into the cluster geometries. This implies that to date there is limited information about how accurately the applied computational methods depict the cluster structures. Here we massively benchmark the molecular geometries of atmospheric molecular clusters. We initially assessed how well different DF-MP2 approaches reproduce the geometries of 45 dimer clusters obtained at a high DF-CCSD(T)-F12b/cc-pVDZ-F12 level of theory. Based on the results, we find that the DF-MP2/aug-cc-pVQZ level of theory best resembles the DF-CCSD(T)-F12b/cc-pVDZ-F12 reference level. We subsequently optimized 1283 acid–base cluster structures (up to tetramers) at the DF-MP2/aug-cc-pVQZ level of theory and assessed how more approximate methods reproduce the geometries. Out of the tested semiempirical methods, we find that the newly parametrized atmospheric molecular cluster extended tight binding method (AMC-xTB) is most reliable for locating the correct lowest energy configuration and yields the lowest root mean square deviation (RMSD) compared to the reference level. In addition, we find that the DFT-3c methods show similar performance as the usually employed ωB97X-D/6-31++G(d,p) level of theory at a potentially reduced computational cost. This suggests that these methods could prove to be valuable for large-scale screening of cluster structures in the future.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
