Andor Menczer, Maarten van Damme, Alan Rask, Lee Huntington, Jeff Hammond, Sotiris S. Xantheas, Martin Ganahl, Örs Legeza
{"title":"在单个 DGX-H100 GPU 节点上实现四分之一 petaFLOPS 性能的密度矩阵重正化群方法并行实施","authors":"Andor Menczer, Maarten van Damme, Alan Rask, Lee Huntington, Jeff Hammond, Sotiris S. Xantheas, Martin Ganahl, Örs Legeza","doi":"10.1021/acs.jctc.4c00903","DOIUrl":null,"url":null,"abstract":"We report cutting edge performance results on a single node hybrid CPU-multi-GPU implementation of the spin adapted <i>ab initio</i> Density Matrix Renormalization Group (DMRG) method on current state-of-the-art NVIDIA DGX-H100 architectures. We evaluate the performance of the DMRG electronic structure calculations for the active compounds of the FeMoco, the primary cofactor of nitrogenase, and cytochrome P450 (CYP) enzymes with complete active space (CAS) sizes of up to 113 electrons in 76 orbitals [CAS(113, 76)] and 63 electrons in 58 orbitals [CAS(63, 58)], respectively. We achieve 246 teraFLOPS of sustained performance, an improvement of more than 2.5× compared to the performance achieved on the DGX-A100 architectures and an 80× acceleration compared to an OpenMP parallelized implementation on a 128-core CPU architecture. Our work highlights the ability of tensor network algorithms to efficiently utilize high-performance multi-GPU hardware and shows that the combination of tensor networks with modern large-scale GPU accelerators can pave the way toward solving some of the most challenging problems in quantum chemistry and beyond.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"735 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-19","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Parallel Implementation of the Density Matrix Renormalization Group Method Achieving a Quarter petaFLOPS Performance on a Single DGX-H100 GPU Node\",\"authors\":\"Andor Menczer, Maarten van Damme, Alan Rask, Lee Huntington, Jeff Hammond, Sotiris S. Xantheas, Martin Ganahl, Örs Legeza\",\"doi\":\"10.1021/acs.jctc.4c00903\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"We report cutting edge performance results on a single node hybrid CPU-multi-GPU implementation of the spin adapted <i>ab initio</i> Density Matrix Renormalization Group (DMRG) method on current state-of-the-art NVIDIA DGX-H100 architectures. We evaluate the performance of the DMRG electronic structure calculations for the active compounds of the FeMoco, the primary cofactor of nitrogenase, and cytochrome P450 (CYP) enzymes with complete active space (CAS) sizes of up to 113 electrons in 76 orbitals [CAS(113, 76)] and 63 electrons in 58 orbitals [CAS(63, 58)], respectively. We achieve 246 teraFLOPS of sustained performance, an improvement of more than 2.5× compared to the performance achieved on the DGX-A100 architectures and an 80× acceleration compared to an OpenMP parallelized implementation on a 128-core CPU architecture. Our work highlights the ability of tensor network algorithms to efficiently utilize high-performance multi-GPU hardware and shows that the combination of tensor networks with modern large-scale GPU accelerators can pave the way toward solving some of the most challenging problems in quantum chemistry and beyond.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"735 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-19\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00903\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00903","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Parallel Implementation of the Density Matrix Renormalization Group Method Achieving a Quarter petaFLOPS Performance on a Single DGX-H100 GPU Node
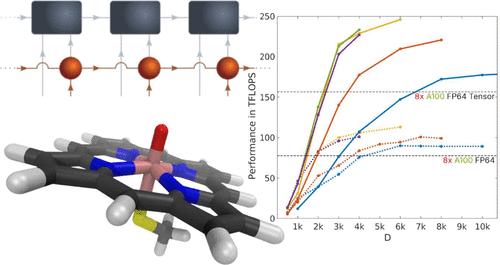
We report cutting edge performance results on a single node hybrid CPU-multi-GPU implementation of the spin adapted ab initio Density Matrix Renormalization Group (DMRG) method on current state-of-the-art NVIDIA DGX-H100 architectures. We evaluate the performance of the DMRG electronic structure calculations for the active compounds of the FeMoco, the primary cofactor of nitrogenase, and cytochrome P450 (CYP) enzymes with complete active space (CAS) sizes of up to 113 electrons in 76 orbitals [CAS(113, 76)] and 63 electrons in 58 orbitals [CAS(63, 58)], respectively. We achieve 246 teraFLOPS of sustained performance, an improvement of more than 2.5× compared to the performance achieved on the DGX-A100 architectures and an 80× acceleration compared to an OpenMP parallelized implementation on a 128-core CPU architecture. Our work highlights the ability of tensor network algorithms to efficiently utilize high-performance multi-GPU hardware and shows that the combination of tensor networks with modern large-scale GPU accelerators can pave the way toward solving some of the most challenging problems in quantum chemistry and beyond.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
