Felipe L. Oliveira, Binquan Luan, Pierre M. Esteves, Mathias Steiner, Rodrigo Neumann Barros Ferreira
{"title":"pyMSER--分子模拟中自动平衡检测的开源库","authors":"Felipe L. Oliveira, Binquan Luan, Pierre M. Esteves, Mathias Steiner, Rodrigo Neumann Barros Ferreira","doi":"10.1021/acs.jctc.4c00417","DOIUrl":null,"url":null,"abstract":"Automated molecular simulations are used extensively for predicting material properties. Typically, these simulations exhibit two regimes: a dynamic equilibration part, followed by a steady state. For extracting observable properties, the simulations must first reach a steady state so that thermodynamic averages can be taken. However, as equilibration depends on simulation conditions, predicting the optimal number of simulation steps <i>a priori</i> is impossible. Here, we demonstrate the application of the Marginal Standard Error Rule (MSER) for automatically identifying the optimal truncation point in Grand Canonical Monte Carlo (GCMC) simulations. This novel automatic procedure determines the point at which a steady state is reached, ensuring that figures of merit are extracted in an objective, accurate, and reproducible fashion. In the case of GCMC simulations of gas adsorption in metal–organic frameworks, we find that this methodology reduces the computational cost by up to 90%. As MSER statistics are independent of the simulation method that creates the data, this library is, in principle, applicable to any time series analysis in which equilibration truncation is required. The open-source Python implementation of our method, <span>pyMSER</span>, is publicly available for reuse and validation at https://github.com/IBM/pymser.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"1 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-18","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"pyMSER─An Open-Source Library for Automatic Equilibration Detection in Molecular Simulations\",\"authors\":\"Felipe L. Oliveira, Binquan Luan, Pierre M. Esteves, Mathias Steiner, Rodrigo Neumann Barros Ferreira\",\"doi\":\"10.1021/acs.jctc.4c00417\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Automated molecular simulations are used extensively for predicting material properties. Typically, these simulations exhibit two regimes: a dynamic equilibration part, followed by a steady state. For extracting observable properties, the simulations must first reach a steady state so that thermodynamic averages can be taken. However, as equilibration depends on simulation conditions, predicting the optimal number of simulation steps <i>a priori</i> is impossible. Here, we demonstrate the application of the Marginal Standard Error Rule (MSER) for automatically identifying the optimal truncation point in Grand Canonical Monte Carlo (GCMC) simulations. This novel automatic procedure determines the point at which a steady state is reached, ensuring that figures of merit are extracted in an objective, accurate, and reproducible fashion. In the case of GCMC simulations of gas adsorption in metal–organic frameworks, we find that this methodology reduces the computational cost by up to 90%. As MSER statistics are independent of the simulation method that creates the data, this library is, in principle, applicable to any time series analysis in which equilibration truncation is required. The open-source Python implementation of our method, <span>pyMSER</span>, is publicly available for reuse and validation at https://github.com/IBM/pymser.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"1 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-18\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00417\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00417","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
pyMSER─An Open-Source Library for Automatic Equilibration Detection in Molecular Simulations
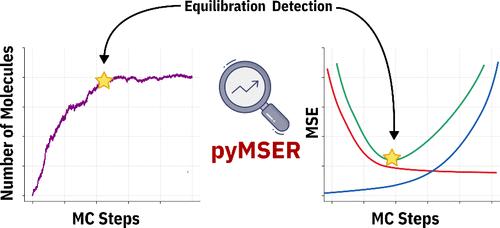
Automated molecular simulations are used extensively for predicting material properties. Typically, these simulations exhibit two regimes: a dynamic equilibration part, followed by a steady state. For extracting observable properties, the simulations must first reach a steady state so that thermodynamic averages can be taken. However, as equilibration depends on simulation conditions, predicting the optimal number of simulation steps a priori is impossible. Here, we demonstrate the application of the Marginal Standard Error Rule (MSER) for automatically identifying the optimal truncation point in Grand Canonical Monte Carlo (GCMC) simulations. This novel automatic procedure determines the point at which a steady state is reached, ensuring that figures of merit are extracted in an objective, accurate, and reproducible fashion. In the case of GCMC simulations of gas adsorption in metal–organic frameworks, we find that this methodology reduces the computational cost by up to 90%. As MSER statistics are independent of the simulation method that creates the data, this library is, in principle, applicable to any time series analysis in which equilibration truncation is required. The open-source Python implementation of our method, pyMSER, is publicly available for reuse and validation at https://github.com/IBM/pymser.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
