Saheli Chowdhury, Abhishek Sen, Debajyoti Das, Partha Chakrabarti
{"title":"去泛素化酶 JOSD1 可调节肝脏蛋白毒性","authors":"Saheli Chowdhury, Abhishek Sen, Debajyoti Das, Partha Chakrabarti","doi":"10.1038/s41420-024-02177-y","DOIUrl":null,"url":null,"abstract":"<p>Derangements in protein homeostasis and associated proteotoxicity mark acute, chronic, and drug-induced hepatocellular injury. Metabolic dysfunction-associated proteasomal inhibition and the use of proteasome inhibitors often underlie such pathological hepatic proteotoxicity. In this study, we sought to identify a candidate deubiquitinating enzyme (DUB) responsible for reversing the proteotoxic damage. To this end, we performed a siRNA screening wherein 96 DUBs were individually knocked down in HepG2 cells under proteasomal inhibitor-induced stress for dual readouts, apoptosis, and cell viability. Among the putative hits, we chose JOSD1, a member of the Machado-Josephin family of DUBs that reciprocally increased cell viability and decreased cell death under proteotoxicity. JOSD1-mediated mitigation of proteotoxicity was further validated in primary mouse hepatocytes by gain and loss of function studies. Marked plasma membrane accumulation of monoubiquitinated JOSD1 in proteotoxic conditions is a prerequisite for its protective role, while the enzymatically inactive JOSD1 C36A mutant was conversely polyubiquitinated, does not have membrane localisation and fails to reverse proteotoxicity. Mechanistically, JOSD1 physically interacts with the suppressor of cytokine signalling 1 (SOCS1), deubiquitinates it and enhances its stability under proteotoxic stress. Indeed, SOCS1 expression is necessary and sufficient for the hepatoprotective function of JOSD1 under proteasomal inhibition. In vivo, adenovirus-mediated ectopic expression or depletion of JOSD1 in mice liver respectively protects or aggravates hepatic injury when challenged with proteasome blocker Bortezomib. Our study thus unveils JOSD1 as a potential candidate for ameliorating hepatocellular damage in liver diseases.</p>","PeriodicalId":9735,"journal":{"name":"Cell Death Discovery","volume":"52 1","pages":""},"PeriodicalIF":7.0000,"publicationDate":"2024-09-16","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Deubiquitinase JOSD1 tempers hepatic proteotoxicity\",\"authors\":\"Saheli Chowdhury, Abhishek Sen, Debajyoti Das, Partha Chakrabarti\",\"doi\":\"10.1038/s41420-024-02177-y\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Derangements in protein homeostasis and associated proteotoxicity mark acute, chronic, and drug-induced hepatocellular injury. Metabolic dysfunction-associated proteasomal inhibition and the use of proteasome inhibitors often underlie such pathological hepatic proteotoxicity. In this study, we sought to identify a candidate deubiquitinating enzyme (DUB) responsible for reversing the proteotoxic damage. To this end, we performed a siRNA screening wherein 96 DUBs were individually knocked down in HepG2 cells under proteasomal inhibitor-induced stress for dual readouts, apoptosis, and cell viability. Among the putative hits, we chose JOSD1, a member of the Machado-Josephin family of DUBs that reciprocally increased cell viability and decreased cell death under proteotoxicity. JOSD1-mediated mitigation of proteotoxicity was further validated in primary mouse hepatocytes by gain and loss of function studies. Marked plasma membrane accumulation of monoubiquitinated JOSD1 in proteotoxic conditions is a prerequisite for its protective role, while the enzymatically inactive JOSD1 C36A mutant was conversely polyubiquitinated, does not have membrane localisation and fails to reverse proteotoxicity. Mechanistically, JOSD1 physically interacts with the suppressor of cytokine signalling 1 (SOCS1), deubiquitinates it and enhances its stability under proteotoxic stress. Indeed, SOCS1 expression is necessary and sufficient for the hepatoprotective function of JOSD1 under proteasomal inhibition. In vivo, adenovirus-mediated ectopic expression or depletion of JOSD1 in mice liver respectively protects or aggravates hepatic injury when challenged with proteasome blocker Bortezomib. Our study thus unveils JOSD1 as a potential candidate for ameliorating hepatocellular damage in liver diseases.</p>\",\"PeriodicalId\":9735,\"journal\":{\"name\":\"Cell Death Discovery\",\"volume\":\"52 1\",\"pages\":\"\"},\"PeriodicalIF\":7.0000,\"publicationDate\":\"2024-09-16\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cell Death Discovery\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41420-024-02177-y\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cell Death Discovery","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41420-024-02177-y","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
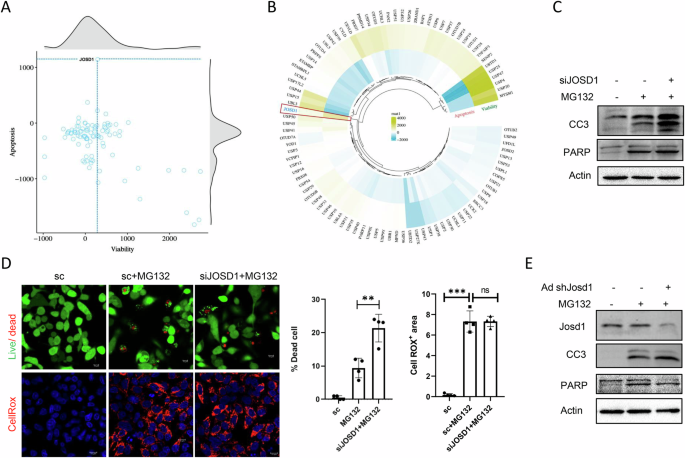
Derangements in protein homeostasis and associated proteotoxicity mark acute, chronic, and drug-induced hepatocellular injury. Metabolic dysfunction-associated proteasomal inhibition and the use of proteasome inhibitors often underlie such pathological hepatic proteotoxicity. In this study, we sought to identify a candidate deubiquitinating enzyme (DUB) responsible for reversing the proteotoxic damage. To this end, we performed a siRNA screening wherein 96 DUBs were individually knocked down in HepG2 cells under proteasomal inhibitor-induced stress for dual readouts, apoptosis, and cell viability. Among the putative hits, we chose JOSD1, a member of the Machado-Josephin family of DUBs that reciprocally increased cell viability and decreased cell death under proteotoxicity. JOSD1-mediated mitigation of proteotoxicity was further validated in primary mouse hepatocytes by gain and loss of function studies. Marked plasma membrane accumulation of monoubiquitinated JOSD1 in proteotoxic conditions is a prerequisite for its protective role, while the enzymatically inactive JOSD1 C36A mutant was conversely polyubiquitinated, does not have membrane localisation and fails to reverse proteotoxicity. Mechanistically, JOSD1 physically interacts with the suppressor of cytokine signalling 1 (SOCS1), deubiquitinates it and enhances its stability under proteotoxic stress. Indeed, SOCS1 expression is necessary and sufficient for the hepatoprotective function of JOSD1 under proteasomal inhibition. In vivo, adenovirus-mediated ectopic expression or depletion of JOSD1 in mice liver respectively protects or aggravates hepatic injury when challenged with proteasome blocker Bortezomib. Our study thus unveils JOSD1 as a potential candidate for ameliorating hepatocellular damage in liver diseases.
期刊介绍:
Cell Death Discovery is a multidisciplinary, international, online-only, open access journal, dedicated to publishing research at the intersection of medicine with biochemistry, pharmacology, immunology, cell biology and cell death, provided it is scientifically sound. The unrestricted access to research findings in Cell Death Discovery will foster a dynamic and highly productive dialogue between basic scientists and clinicians, as well as researchers in industry with a focus on cancer, neurobiology and inflammation research. As an official journal of the Cell Death Differentiation Association (ADMC), Cell Death Discovery will build upon the success of Cell Death & Differentiation and Cell Death & Disease in publishing important peer-reviewed original research, timely reviews and editorial commentary.
Cell Death Discovery is committed to increasing the reproducibility of research. To this end, in conjunction with its sister journals Cell Death & Differentiation and Cell Death & Disease, Cell Death Discovery provides a unique forum for scientists as well as clinicians and members of the pharmaceutical and biotechnical industry. It is committed to the rapid publication of high quality original papers that relate to these subjects, together with topical, usually solicited, reviews, editorial correspondence and occasional commentaries on controversial and scientifically informative issues.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
