Ieva Liepuoniute, Mario Motta, Thaddeus Pellegrini, Julia E. Rice, Tanvi P. Gujarati, Sofia Gil and Gavin O. Jones
{"title":"在量子计算机上模拟 Diels-Alder 反应。","authors":"Ieva Liepuoniute, Mario Motta, Thaddeus Pellegrini, Julia E. Rice, Tanvi P. Gujarati, Sofia Gil and Gavin O. Jones","doi":"10.1039/D4CP01314J","DOIUrl":null,"url":null,"abstract":"<p >The simulation of chemical reactions is an anticipated application of quantum computers. Using a Diels–Alder reaction as a test case, in this study we explore the potential applications of quantum algorithms and hardware in investigating chemical reactions. Our specific goal is to calculate the activation barrier of a reaction between ethylene and cyclopentadiene forming a transition state. To achieve this goal, we use quantum algorithms for near-term quantum hardware (entanglement forging and quantum subspace expansion) and classical post-processing (many-body perturbation theory) in concert. We conduct simulations on IBM quantum hardware using up to 8 qubits, and compute accurate activation barrier in the reaction between cyclopentadiene and ethylene by accounting for both static and dynamic electronic correlation. This work illustrates a hybrid quantum-classical computational workflow to study chemical reactions on near-term quantum devices, showcasing the potential for performing quantum chemistry simulations on quantum hardware to predict activation barriers in agreement with those predicted by CASCI.</p>","PeriodicalId":99,"journal":{"name":"Physical Chemistry Chemical Physics","volume":" 38","pages":" 25181-25191"},"PeriodicalIF":2.9000,"publicationDate":"2024-09-24","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp01314j?page=search","citationCount":"0","resultStr":"{\"title\":\"Simulation of a Diels–Alder reaction on a quantum computer\",\"authors\":\"Ieva Liepuoniute, Mario Motta, Thaddeus Pellegrini, Julia E. Rice, Tanvi P. Gujarati, Sofia Gil and Gavin O. Jones\",\"doi\":\"10.1039/D4CP01314J\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p >The simulation of chemical reactions is an anticipated application of quantum computers. Using a Diels–Alder reaction as a test case, in this study we explore the potential applications of quantum algorithms and hardware in investigating chemical reactions. Our specific goal is to calculate the activation barrier of a reaction between ethylene and cyclopentadiene forming a transition state. To achieve this goal, we use quantum algorithms for near-term quantum hardware (entanglement forging and quantum subspace expansion) and classical post-processing (many-body perturbation theory) in concert. We conduct simulations on IBM quantum hardware using up to 8 qubits, and compute accurate activation barrier in the reaction between cyclopentadiene and ethylene by accounting for both static and dynamic electronic correlation. This work illustrates a hybrid quantum-classical computational workflow to study chemical reactions on near-term quantum devices, showcasing the potential for performing quantum chemistry simulations on quantum hardware to predict activation barriers in agreement with those predicted by CASCI.</p>\",\"PeriodicalId\":99,\"journal\":{\"name\":\"Physical Chemistry Chemical Physics\",\"volume\":\" 38\",\"pages\":\" 25181-25191\"},\"PeriodicalIF\":2.9000,\"publicationDate\":\"2024-09-24\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://pubs.rsc.org/en/content/articlepdf/2024/cp/d4cp01314j?page=search\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Physical Chemistry Chemical Physics\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01314j\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Physical Chemistry Chemical Physics","FirstCategoryId":"92","ListUrlMain":"https://pubs.rsc.org/en/content/articlelanding/2024/cp/d4cp01314j","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
模拟化学反应是量子计算机的一项预期应用。本研究以 Diels-Alder 反应为测试案例,探索量子算法和硬件在化学反应研究中的潜在应用。我们的具体目标是计算乙烯和环戊二烯形成过渡态反应的活化势垒。为了实现这一目标,我们将近期量子硬件的量子算法(纠缠锻造和量子子空间扩展)和经典后处理(多体扰动理论)结合起来使用。我们在 IBM 量子硬件上使用多达 8 个量子比特进行了模拟,并通过考虑静态和动态电子相关计算出了环戊二烯和乙烯反应的精确活化势垒。这项工作说明了在近期量子设备上研究化学反应的混合量子-经典计算工作流程,展示了在量子硬件上进行量子化学模拟以预测与 CASCI 预测一致的活化势垒的潜力。
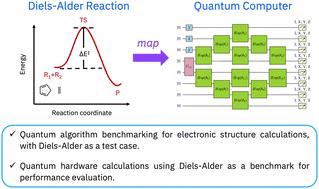
Simulation of a Diels–Alder reaction on a quantum computer
The simulation of chemical reactions is an anticipated application of quantum computers. Using a Diels–Alder reaction as a test case, in this study we explore the potential applications of quantum algorithms and hardware in investigating chemical reactions. Our specific goal is to calculate the activation barrier of a reaction between ethylene and cyclopentadiene forming a transition state. To achieve this goal, we use quantum algorithms for near-term quantum hardware (entanglement forging and quantum subspace expansion) and classical post-processing (many-body perturbation theory) in concert. We conduct simulations on IBM quantum hardware using up to 8 qubits, and compute accurate activation barrier in the reaction between cyclopentadiene and ethylene by accounting for both static and dynamic electronic correlation. This work illustrates a hybrid quantum-classical computational workflow to study chemical reactions on near-term quantum devices, showcasing the potential for performing quantum chemistry simulations on quantum hardware to predict activation barriers in agreement with those predicted by CASCI.
期刊介绍:
Physical Chemistry Chemical Physics (PCCP) is an international journal co-owned by 19 physical chemistry and physics societies from around the world. This journal publishes original, cutting-edge research in physical chemistry, chemical physics and biophysical chemistry. To be suitable for publication in PCCP, articles must include significant innovation and/or insight into physical chemistry; this is the most important criterion that reviewers and Editors will judge against when evaluating submissions.
The journal has a broad scope and welcomes contributions spanning experiment, theory, computation and data science. Topical coverage includes spectroscopy, dynamics, kinetics, statistical mechanics, thermodynamics, electrochemistry, catalysis, surface science, quantum mechanics, quantum computing and machine learning. Interdisciplinary research areas such as polymers and soft matter, materials, nanoscience, energy, surfaces/interfaces, and biophysical chemistry are welcomed if they demonstrate significant innovation and/or insight into physical chemistry. Joined experimental/theoretical studies are particularly appreciated when complementary and based on up-to-date approaches.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
