Jan K. Dabrowski, Emma J. Yang, Samuel J. C. Crofts, Robert F. Hillary, Daniel J. Simpson, Daniel L. McCartney, Riccardo E. Marioni, Kristina Kirschner, Eric Latorre-Crespo, Tamir Chandra
{"title":"从细胞动力学推断表观遗传年龄加速的概率。","authors":"Jan K. Dabrowski, Emma J. Yang, Samuel J. C. Crofts, Robert F. Hillary, Daniel J. Simpson, Daniel L. McCartney, Riccardo E. Marioni, Kristina Kirschner, Eric Latorre-Crespo, Tamir Chandra","doi":"10.1038/s43587-024-00700-5","DOIUrl":null,"url":null,"abstract":"The emergence of epigenetic predictors was a pivotal moment in geroscience, propelling the measurement and concept of biological aging into a quantitative era; however, while current epigenetic clocks show strong predictive power, they are data-driven in nature and are not based on the underlying biological mechanisms driving methylation dynamics. We show that predictions of these clocks are susceptible to several confounding non-age-related phenomena that make interpretation of these estimates and associations difficult. To address these limitations, we developed a probabilistic model describing methylation transitions at the cellular level. Our approach reveals two measurable components, acceleration and bias, which directly reflect perturbations of the underlying cellular dynamics. Acceleration is the proportional increase in the speed of methylation transitions across CpG sites, whereas bias corresponds to global changes in methylation levels. Using data from 15,900 participants from the Generation Scotland study, we develop a robust inference framework and show that these are two distinct processes confounding current epigenetic predictors. Our results show improved associations of acceleration and bias with physiological traits known to impact healthy aging, such as smoking and alcohol consumption, respectively. Furthermore, a genome-wide association study of epigenetic age acceleration identified seven genomic loci. Epigenetic clocks estimate biological age; however, the underlying processes are incompletely understood. Here the authors developed a model that describes methylation changes at the cellular level to provide a biologically interpretable predictor of epigenetic age. They reveal acceleration and bias components, and links with health traits.","PeriodicalId":94150,"journal":{"name":"Nature aging","volume":"4 10","pages":"1493-1507"},"PeriodicalIF":19.4000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s43587-024-00700-5.pdf","citationCount":"0","resultStr":"{\"title\":\"Probabilistic inference of epigenetic age acceleration from cellular dynamics\",\"authors\":\"Jan K. Dabrowski, Emma J. Yang, Samuel J. C. Crofts, Robert F. Hillary, Daniel J. Simpson, Daniel L. McCartney, Riccardo E. Marioni, Kristina Kirschner, Eric Latorre-Crespo, Tamir Chandra\",\"doi\":\"10.1038/s43587-024-00700-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The emergence of epigenetic predictors was a pivotal moment in geroscience, propelling the measurement and concept of biological aging into a quantitative era; however, while current epigenetic clocks show strong predictive power, they are data-driven in nature and are not based on the underlying biological mechanisms driving methylation dynamics. We show that predictions of these clocks are susceptible to several confounding non-age-related phenomena that make interpretation of these estimates and associations difficult. To address these limitations, we developed a probabilistic model describing methylation transitions at the cellular level. Our approach reveals two measurable components, acceleration and bias, which directly reflect perturbations of the underlying cellular dynamics. Acceleration is the proportional increase in the speed of methylation transitions across CpG sites, whereas bias corresponds to global changes in methylation levels. Using data from 15,900 participants from the Generation Scotland study, we develop a robust inference framework and show that these are two distinct processes confounding current epigenetic predictors. Our results show improved associations of acceleration and bias with physiological traits known to impact healthy aging, such as smoking and alcohol consumption, respectively. Furthermore, a genome-wide association study of epigenetic age acceleration identified seven genomic loci. Epigenetic clocks estimate biological age; however, the underlying processes are incompletely understood. Here the authors developed a model that describes methylation changes at the cellular level to provide a biologically interpretable predictor of epigenetic age. They reveal acceleration and bias components, and links with health traits.\",\"PeriodicalId\":94150,\"journal\":{\"name\":\"Nature aging\",\"volume\":\"4 10\",\"pages\":\"1493-1507\"},\"PeriodicalIF\":19.4000,\"publicationDate\":\"2024-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s43587-024-00700-5.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature aging\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://www.nature.com/articles/s43587-024-00700-5\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature aging","FirstCategoryId":"1085","ListUrlMain":"https://www.nature.com/articles/s43587-024-00700-5","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Probabilistic inference of epigenetic age acceleration from cellular dynamics
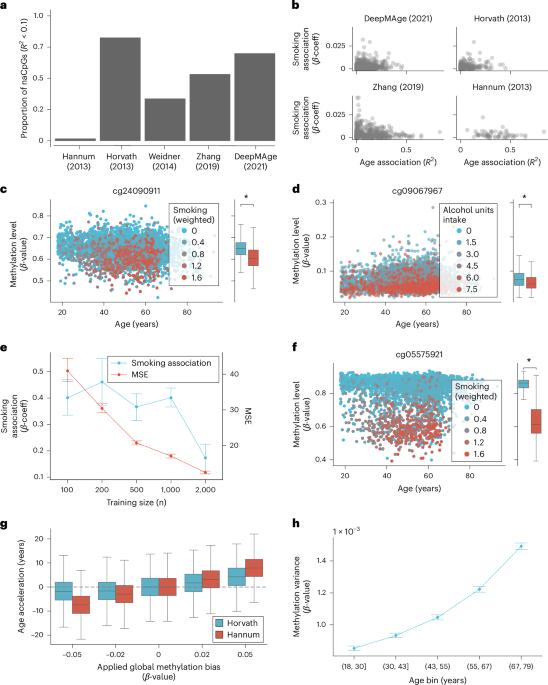
The emergence of epigenetic predictors was a pivotal moment in geroscience, propelling the measurement and concept of biological aging into a quantitative era; however, while current epigenetic clocks show strong predictive power, they are data-driven in nature and are not based on the underlying biological mechanisms driving methylation dynamics. We show that predictions of these clocks are susceptible to several confounding non-age-related phenomena that make interpretation of these estimates and associations difficult. To address these limitations, we developed a probabilistic model describing methylation transitions at the cellular level. Our approach reveals two measurable components, acceleration and bias, which directly reflect perturbations of the underlying cellular dynamics. Acceleration is the proportional increase in the speed of methylation transitions across CpG sites, whereas bias corresponds to global changes in methylation levels. Using data from 15,900 participants from the Generation Scotland study, we develop a robust inference framework and show that these are two distinct processes confounding current epigenetic predictors. Our results show improved associations of acceleration and bias with physiological traits known to impact healthy aging, such as smoking and alcohol consumption, respectively. Furthermore, a genome-wide association study of epigenetic age acceleration identified seven genomic loci. Epigenetic clocks estimate biological age; however, the underlying processes are incompletely understood. Here the authors developed a model that describes methylation changes at the cellular level to provide a biologically interpretable predictor of epigenetic age. They reveal acceleration and bias components, and links with health traits.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
