{"title":"对 M-Salen 和 M-Salphen 电催化剂的电子和结构特性进行 DFT 研究,以实现有效的 HER","authors":"Saravanapriya Arumugam , Abiram Angamuthu , Praveena Gopalan","doi":"10.1016/j.jics.2024.101381","DOIUrl":null,"url":null,"abstract":"<div><div>We report on the electrochemical properties of Salen (<em>N, N’- bis salicylaldehyde ehtylenediamine</em>) and Salphen (<em>N, N’- bis salicylaldehyde phenylenediamine</em>) ligands using density functional approach. The structural and electronic properties, and the reduction potentials of metalated M-Salen and M-Salphen ligands (where M = Sb & Mo) that involve in hydrogen evolution reaction were explored. Optimized geometries of the chosen metalated complexes were obtained at B3LYP/6-31+G(d, p) & LANL2DZ level of theory. The effects solvation on the electrochemical properties of M-Salen and M-Salphen systems were considered in the presence of solvent acetonitrile using conductor-like polarisable continuum model (CPCM) at the same level of theory. Upon reduction process, the charge distribution around the metal centers Mo and Sb, and C, N and O atoms that lie in the coordination sphere is found to change considerably. As the first unoccupied orbital LUMO is directly connected to the electron affinity, the greater negative values of LUMO observed in Mo substituted Salen and Salphen ligands indicate their ability to exhibit better reduction process. Calculated reduction potential values of M-Salen systems were found to vary from −2.23V to −0.62V and hence the catalytic activity of M-Salen ligands follows the order of Mo-Salen > Sb-Salen > Salen and the same trend has been observed in M-Salphen systems with enhanced reduction potential of −0.54V recorded for Mo-Salphen system.</div></div>","PeriodicalId":17276,"journal":{"name":"Journal of the Indian Chemical Society","volume":"101 11","pages":"Article 101381"},"PeriodicalIF":3.2000,"publicationDate":"2024-09-17","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"DFT study on the electronic and structural properties of M-Salen and M-Salphen electrocatalysts towards effective HER\",\"authors\":\"Saravanapriya Arumugam , Abiram Angamuthu , Praveena Gopalan\",\"doi\":\"10.1016/j.jics.2024.101381\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>We report on the electrochemical properties of Salen (<em>N, N’- bis salicylaldehyde ehtylenediamine</em>) and Salphen (<em>N, N’- bis salicylaldehyde phenylenediamine</em>) ligands using density functional approach. The structural and electronic properties, and the reduction potentials of metalated M-Salen and M-Salphen ligands (where M = Sb & Mo) that involve in hydrogen evolution reaction were explored. Optimized geometries of the chosen metalated complexes were obtained at B3LYP/6-31+G(d, p) & LANL2DZ level of theory. The effects solvation on the electrochemical properties of M-Salen and M-Salphen systems were considered in the presence of solvent acetonitrile using conductor-like polarisable continuum model (CPCM) at the same level of theory. Upon reduction process, the charge distribution around the metal centers Mo and Sb, and C, N and O atoms that lie in the coordination sphere is found to change considerably. As the first unoccupied orbital LUMO is directly connected to the electron affinity, the greater negative values of LUMO observed in Mo substituted Salen and Salphen ligands indicate their ability to exhibit better reduction process. Calculated reduction potential values of M-Salen systems were found to vary from −2.23V to −0.62V and hence the catalytic activity of M-Salen ligands follows the order of Mo-Salen > Sb-Salen > Salen and the same trend has been observed in M-Salphen systems with enhanced reduction potential of −0.54V recorded for Mo-Salphen system.</div></div>\",\"PeriodicalId\":17276,\"journal\":{\"name\":\"Journal of the Indian Chemical Society\",\"volume\":\"101 11\",\"pages\":\"Article 101381\"},\"PeriodicalIF\":3.2000,\"publicationDate\":\"2024-09-17\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of the Indian Chemical Society\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0019452224002619\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of the Indian Chemical Society","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0019452224002619","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
DFT study on the electronic and structural properties of M-Salen and M-Salphen electrocatalysts towards effective HER
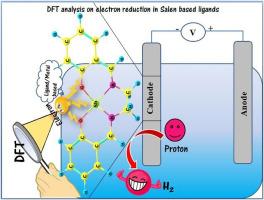
We report on the electrochemical properties of Salen (N, N’- bis salicylaldehyde ehtylenediamine) and Salphen (N, N’- bis salicylaldehyde phenylenediamine) ligands using density functional approach. The structural and electronic properties, and the reduction potentials of metalated M-Salen and M-Salphen ligands (where M = Sb & Mo) that involve in hydrogen evolution reaction were explored. Optimized geometries of the chosen metalated complexes were obtained at B3LYP/6-31+G(d, p) & LANL2DZ level of theory. The effects solvation on the electrochemical properties of M-Salen and M-Salphen systems were considered in the presence of solvent acetonitrile using conductor-like polarisable continuum model (CPCM) at the same level of theory. Upon reduction process, the charge distribution around the metal centers Mo and Sb, and C, N and O atoms that lie in the coordination sphere is found to change considerably. As the first unoccupied orbital LUMO is directly connected to the electron affinity, the greater negative values of LUMO observed in Mo substituted Salen and Salphen ligands indicate their ability to exhibit better reduction process. Calculated reduction potential values of M-Salen systems were found to vary from −2.23V to −0.62V and hence the catalytic activity of M-Salen ligands follows the order of Mo-Salen > Sb-Salen > Salen and the same trend has been observed in M-Salphen systems with enhanced reduction potential of −0.54V recorded for Mo-Salphen system.
期刊介绍:
The Journal of the Indian Chemical Society publishes original, fundamental, theorical, experimental research work of highest quality in all areas of chemistry, biochemistry, medicinal chemistry, electrochemistry, agrochemistry, chemical engineering and technology, food chemistry, environmental chemistry, etc.


 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
