Sheila M. Gaynor, Tyler Joseph, Xiaodong Bai, Yuxin Zou, Boris Boutkov, Evan K. Maxwell, Olivier Delaneau, Robin J. Hofmeister, Olga Krasheninina, Suganthi Balasubramanian, Anthony Marcketta, Joshua Backman, Regeneron Genetics Center, Jeffrey G. Reid, John D. Overton, Luca A. Lotta, Jonathan Marchini, William J. Salerno, Aris Baras, Goncalo R. Abecasis, Timothy A. Thornton
{"title":"英国生物库中基因组、外显子组和估算的遗传关联信号的产量","authors":"Sheila M. Gaynor, Tyler Joseph, Xiaodong Bai, Yuxin Zou, Boris Boutkov, Evan K. Maxwell, Olivier Delaneau, Robin J. Hofmeister, Olga Krasheninina, Suganthi Balasubramanian, Anthony Marcketta, Joshua Backman, Regeneron Genetics Center, Jeffrey G. Reid, John D. Overton, Luca A. Lotta, Jonathan Marchini, William J. Salerno, Aris Baras, Goncalo R. Abecasis, Timothy A. Thornton","doi":"10.1038/s41588-024-01930-4","DOIUrl":null,"url":null,"abstract":"Whole-genome sequencing (WGS), whole-exome sequencing (WES) and array genotyping with imputation (IMP) are common strategies for assessing genetic variation and its association with medically relevant phenotypes. To date, there has been no systematic empirical assessment of the yield of these approaches when applied to hundreds of thousands of samples to enable the discovery of complex trait genetic signals. Using data for 100 complex traits from 149,195 individuals in the UK Biobank, we systematically compare the relative yield of these strategies in genetic association studies. We find that WGS and WES combined with arrays and imputation (WES + IMP) have the largest association yield. Although WGS results in an approximately fivefold increase in the total number of assayed variants over WES + IMP, the number of detected signals differed by only 1% for both single-variant and gene-based association analyses. Given that WES + IMP typically results in savings of lab and computational time and resources expended per sample, we evaluate the potential benefits of applying WES + IMP to larger samples. When we extend our WES + IMP analyses to 468,169 UK Biobank individuals, we observe an approximately fourfold increase in association signals with the threefold increase in sample size. We conclude that prioritizing WES + IMP and large sample sizes rather than contemporary short-read WGS alternatives will maximize the number of discoveries in genetic association studies. Comparison of association signals in UK Biobank using different strategies for assessing genetic variation shows that whole-exome sequencing combined with array genotyping and imputation offers similar performance to whole-genome sequencing at a reduced cost.","PeriodicalId":18985,"journal":{"name":"Nature genetics","volume":"56 11","pages":"2345-2351"},"PeriodicalIF":25.5000,"publicationDate":"2024-09-25","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.nature.com/articles/s41588-024-01930-4.pdf","citationCount":"0","resultStr":"{\"title\":\"Yield of genetic association signals from genomes, exomes and imputation in the UK Biobank\",\"authors\":\"Sheila M. Gaynor, Tyler Joseph, Xiaodong Bai, Yuxin Zou, Boris Boutkov, Evan K. Maxwell, Olivier Delaneau, Robin J. Hofmeister, Olga Krasheninina, Suganthi Balasubramanian, Anthony Marcketta, Joshua Backman, Regeneron Genetics Center, Jeffrey G. Reid, John D. Overton, Luca A. Lotta, Jonathan Marchini, William J. Salerno, Aris Baras, Goncalo R. Abecasis, Timothy A. Thornton\",\"doi\":\"10.1038/s41588-024-01930-4\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Whole-genome sequencing (WGS), whole-exome sequencing (WES) and array genotyping with imputation (IMP) are common strategies for assessing genetic variation and its association with medically relevant phenotypes. To date, there has been no systematic empirical assessment of the yield of these approaches when applied to hundreds of thousands of samples to enable the discovery of complex trait genetic signals. Using data for 100 complex traits from 149,195 individuals in the UK Biobank, we systematically compare the relative yield of these strategies in genetic association studies. We find that WGS and WES combined with arrays and imputation (WES + IMP) have the largest association yield. Although WGS results in an approximately fivefold increase in the total number of assayed variants over WES + IMP, the number of detected signals differed by only 1% for both single-variant and gene-based association analyses. Given that WES + IMP typically results in savings of lab and computational time and resources expended per sample, we evaluate the potential benefits of applying WES + IMP to larger samples. When we extend our WES + IMP analyses to 468,169 UK Biobank individuals, we observe an approximately fourfold increase in association signals with the threefold increase in sample size. We conclude that prioritizing WES + IMP and large sample sizes rather than contemporary short-read WGS alternatives will maximize the number of discoveries in genetic association studies. Comparison of association signals in UK Biobank using different strategies for assessing genetic variation shows that whole-exome sequencing combined with array genotyping and imputation offers similar performance to whole-genome sequencing at a reduced cost.\",\"PeriodicalId\":18985,\"journal\":{\"name\":\"Nature genetics\",\"volume\":\"56 11\",\"pages\":\"2345-2351\"},\"PeriodicalIF\":25.5000,\"publicationDate\":\"2024-09-25\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.nature.com/articles/s41588-024-01930-4.pdf\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Nature genetics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.nature.com/articles/s41588-024-01930-4\",\"RegionNum\":1,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Nature genetics","FirstCategoryId":"99","ListUrlMain":"https://www.nature.com/articles/s41588-024-01930-4","RegionNum":1,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Yield of genetic association signals from genomes, exomes and imputation in the UK Biobank
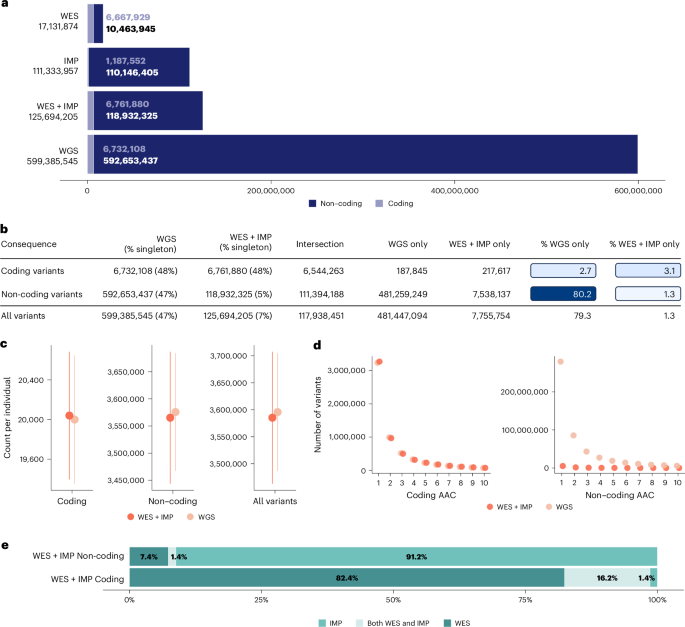
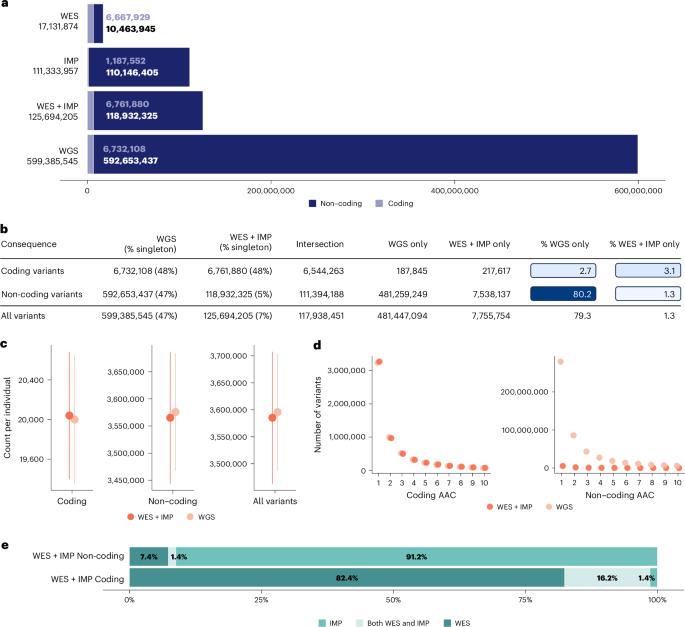
Whole-genome sequencing (WGS), whole-exome sequencing (WES) and array genotyping with imputation (IMP) are common strategies for assessing genetic variation and its association with medically relevant phenotypes. To date, there has been no systematic empirical assessment of the yield of these approaches when applied to hundreds of thousands of samples to enable the discovery of complex trait genetic signals. Using data for 100 complex traits from 149,195 individuals in the UK Biobank, we systematically compare the relative yield of these strategies in genetic association studies. We find that WGS and WES combined with arrays and imputation (WES + IMP) have the largest association yield. Although WGS results in an approximately fivefold increase in the total number of assayed variants over WES + IMP, the number of detected signals differed by only 1% for both single-variant and gene-based association analyses. Given that WES + IMP typically results in savings of lab and computational time and resources expended per sample, we evaluate the potential benefits of applying WES + IMP to larger samples. When we extend our WES + IMP analyses to 468,169 UK Biobank individuals, we observe an approximately fourfold increase in association signals with the threefold increase in sample size. We conclude that prioritizing WES + IMP and large sample sizes rather than contemporary short-read WGS alternatives will maximize the number of discoveries in genetic association studies. Comparison of association signals in UK Biobank using different strategies for assessing genetic variation shows that whole-exome sequencing combined with array genotyping and imputation offers similar performance to whole-genome sequencing at a reduced cost.
期刊介绍:
Nature Genetics publishes the very highest quality research in genetics. It encompasses genetic and functional genomic studies on human and plant traits and on other model organisms. Current emphasis is on the genetic basis for common and complex diseases and on the functional mechanism, architecture and evolution of gene networks, studied by experimental perturbation.
Integrative genetic topics comprise, but are not limited to:
-Genes in the pathology of human disease
-Molecular analysis of simple and complex genetic traits
-Cancer genetics
-Agricultural genomics
-Developmental genetics
-Regulatory variation in gene expression
-Strategies and technologies for extracting function from genomic data
-Pharmacological genomics
-Genome evolution




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
