{"title":"金属间化合物 Fe3Al 的真实基态是什么?","authors":"M. Všianská , M. Šob","doi":"10.1016/j.solidstatesciences.2024.107709","DOIUrl":null,"url":null,"abstract":"<div><div>We discuss recent doubts about the true ground-state (GS) structure of the intermetallic compound Fe<sub>3</sub>Al. It seems that it should be the D0<sub>3</sub> structure (observed experimentally), but there are some considerations that, perhaps, D0<sub>3</sub> might be a high-temperature (>400 K) structure and the GS at 0 K might be the L1<sub>2</sub> structure because there might be a high energy barrier between both structures and, when the temperature is lowered, the system is not able to transform into the (perhaps) lower-energy L1<sub>2</sub> structure. To elucidate this problem, we re-interpret our recent extended ab initio electronic structure calculations for Fe<sub>3</sub>Al performed with the help of the VASP code and using various exchange-correlation energies within the generalized gradient approximation (GGA). Regrettably, some calculations provide the L1<sub>2</sub> and some of them D0<sub>3</sub> as the GS structure.</div><div>To resolve this question, we performed further calculations testing 9 frequently applied metaGGAs, such as TPSS, revTPSS, M06-L, SCAN(-L), rSCAN(-L) and r<sup>2</sup>SCAN(-L) representing a higher rung of the Jacob's ladder. It turns out that also here some meta-GGAs lead to L1<sub>2</sub> and some others to D0<sub>3</sub> GS structure and, again, we cannot decide.</div><div>In this way, the present results represent the very first step on the way to understand the energetics of the Fe<sub>3</sub>Al compound and its ground state. We hope they may motivate future theoretical and experimental work in this direction.</div></div>","PeriodicalId":432,"journal":{"name":"Solid State Sciences","volume":"157 ","pages":"Article 107709"},"PeriodicalIF":3.3000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"What is the true ground state of intermetallic compound Fe3Al?\",\"authors\":\"M. Všianská , M. Šob\",\"doi\":\"10.1016/j.solidstatesciences.2024.107709\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>We discuss recent doubts about the true ground-state (GS) structure of the intermetallic compound Fe<sub>3</sub>Al. It seems that it should be the D0<sub>3</sub> structure (observed experimentally), but there are some considerations that, perhaps, D0<sub>3</sub> might be a high-temperature (>400 K) structure and the GS at 0 K might be the L1<sub>2</sub> structure because there might be a high energy barrier between both structures and, when the temperature is lowered, the system is not able to transform into the (perhaps) lower-energy L1<sub>2</sub> structure. To elucidate this problem, we re-interpret our recent extended ab initio electronic structure calculations for Fe<sub>3</sub>Al performed with the help of the VASP code and using various exchange-correlation energies within the generalized gradient approximation (GGA). Regrettably, some calculations provide the L1<sub>2</sub> and some of them D0<sub>3</sub> as the GS structure.</div><div>To resolve this question, we performed further calculations testing 9 frequently applied metaGGAs, such as TPSS, revTPSS, M06-L, SCAN(-L), rSCAN(-L) and r<sup>2</sup>SCAN(-L) representing a higher rung of the Jacob's ladder. It turns out that also here some meta-GGAs lead to L1<sub>2</sub> and some others to D0<sub>3</sub> GS structure and, again, we cannot decide.</div><div>In this way, the present results represent the very first step on the way to understand the energetics of the Fe<sub>3</sub>Al compound and its ground state. We hope they may motivate future theoretical and experimental work in this direction.</div></div>\",\"PeriodicalId\":432,\"journal\":{\"name\":\"Solid State Sciences\",\"volume\":\"157 \",\"pages\":\"Article 107709\"},\"PeriodicalIF\":3.3000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Solid State Sciences\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S1293255824002747\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/21 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, INORGANIC & NUCLEAR\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Solid State Sciences","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S1293255824002747","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/21 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"CHEMISTRY, INORGANIC & NUCLEAR","Score":null,"Total":0}
What is the true ground state of intermetallic compound Fe3Al?
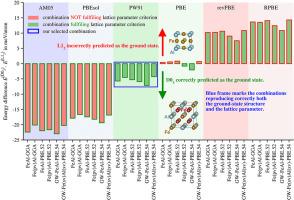
We discuss recent doubts about the true ground-state (GS) structure of the intermetallic compound Fe3Al. It seems that it should be the D03 structure (observed experimentally), but there are some considerations that, perhaps, D03 might be a high-temperature (>400 K) structure and the GS at 0 K might be the L12 structure because there might be a high energy barrier between both structures and, when the temperature is lowered, the system is not able to transform into the (perhaps) lower-energy L12 structure. To elucidate this problem, we re-interpret our recent extended ab initio electronic structure calculations for Fe3Al performed with the help of the VASP code and using various exchange-correlation energies within the generalized gradient approximation (GGA). Regrettably, some calculations provide the L12 and some of them D03 as the GS structure.
To resolve this question, we performed further calculations testing 9 frequently applied metaGGAs, such as TPSS, revTPSS, M06-L, SCAN(-L), rSCAN(-L) and r2SCAN(-L) representing a higher rung of the Jacob's ladder. It turns out that also here some meta-GGAs lead to L12 and some others to D03 GS structure and, again, we cannot decide.
In this way, the present results represent the very first step on the way to understand the energetics of the Fe3Al compound and its ground state. We hope they may motivate future theoretical and experimental work in this direction.
期刊介绍:
Solid State Sciences is the journal for researchers from the broad solid state chemistry and physics community. It publishes key articles on all aspects of solid state synthesis, structure-property relationships, theory and functionalities, in relation with experiments.
Key topics for stand-alone papers and special issues:
-Novel ways of synthesis, inorganic functional materials, including porous and glassy materials, hybrid organic-inorganic compounds and nanomaterials
-Physical properties, emphasizing but not limited to the electrical, magnetical and optical features
-Materials related to information technology and energy and environmental sciences.
The journal publishes feature articles from experts in the field upon invitation.
Solid State Sciences - your gateway to energy-related materials.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
