Jakob R. Riccabona, Fabian C. Spoendlin, Anna-Lena M. Fischer, Johannes R. Loeffler, Patrick K. Quoika, Timothy P. Jenkins, James A. Ferguson, Eva Smorodina, Andreas H. Laustsen, Victor Greiff, Stefano Forli, Andrew B. Ward, Charlotte M. Deane, Monica L. Fernández-Quintero
{"title":"评估 AF2 预测蛋白质结构组合的能力","authors":"Jakob R. Riccabona, Fabian C. Spoendlin, Anna-Lena M. Fischer, Johannes R. Loeffler, Patrick K. Quoika, Timothy P. Jenkins, James A. Ferguson, Eva Smorodina, Andreas H. Laustsen, Victor Greiff, Stefano Forli, Andrew B. Ward, Charlotte M. Deane, Monica L. Fernández-Quintero","doi":"10.1016/j.str.2024.09.001","DOIUrl":null,"url":null,"abstract":"Recent breakthroughs in protein structure prediction have enhanced the precision and speed at which protein configurations can be determined. Additionally, molecular dynamics (MD) simulations serve as a crucial tool for capturing the conformational space of proteins, providing valuable insights into their structural fluctuations. However, the scope of MD simulations is often limited by the accessible timescales and the computational resources available, posing challenges to comprehensively exploring protein behaviors. Recently emerging approaches have focused on expanding the capability of AlphaFold2 (AF2) to predict conformational substates of protein. Here, we benchmark the performance of various workflows that have adapted AF2 for ensemble prediction and compare the obtained structures with ensembles obtained from MD simulations and NMR. We provide an overview of the levels of performance and accessible timescales that can currently be achieved with machine learning (ML) based ensemble generation. Significant minima of the free energy surfaces remain undetected.","PeriodicalId":22168,"journal":{"name":"Structure","volume":"24 1","pages":""},"PeriodicalIF":4.6000,"publicationDate":"2024-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Assessing AF2’s ability to predict structural ensembles of proteins\",\"authors\":\"Jakob R. Riccabona, Fabian C. Spoendlin, Anna-Lena M. Fischer, Johannes R. Loeffler, Patrick K. Quoika, Timothy P. Jenkins, James A. Ferguson, Eva Smorodina, Andreas H. Laustsen, Victor Greiff, Stefano Forli, Andrew B. Ward, Charlotte M. Deane, Monica L. Fernández-Quintero\",\"doi\":\"10.1016/j.str.2024.09.001\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Recent breakthroughs in protein structure prediction have enhanced the precision and speed at which protein configurations can be determined. Additionally, molecular dynamics (MD) simulations serve as a crucial tool for capturing the conformational space of proteins, providing valuable insights into their structural fluctuations. However, the scope of MD simulations is often limited by the accessible timescales and the computational resources available, posing challenges to comprehensively exploring protein behaviors. Recently emerging approaches have focused on expanding the capability of AlphaFold2 (AF2) to predict conformational substates of protein. Here, we benchmark the performance of various workflows that have adapted AF2 for ensemble prediction and compare the obtained structures with ensembles obtained from MD simulations and NMR. We provide an overview of the levels of performance and accessible timescales that can currently be achieved with machine learning (ML) based ensemble generation. Significant minima of the free energy surfaces remain undetected.\",\"PeriodicalId\":22168,\"journal\":{\"name\":\"Structure\",\"volume\":\"24 1\",\"pages\":\"\"},\"PeriodicalIF\":4.6000,\"publicationDate\":\"2024-09-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Structure\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1016/j.str.2024.09.001\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Structure","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1016/j.str.2024.09.001","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
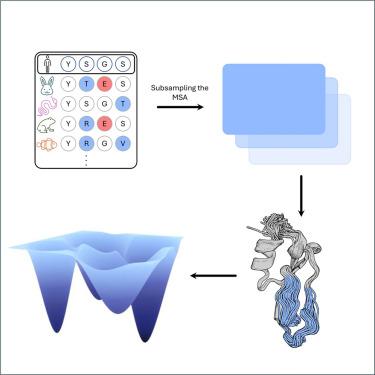
Assessing AF2’s ability to predict structural ensembles of proteins
Recent breakthroughs in protein structure prediction have enhanced the precision and speed at which protein configurations can be determined. Additionally, molecular dynamics (MD) simulations serve as a crucial tool for capturing the conformational space of proteins, providing valuable insights into their structural fluctuations. However, the scope of MD simulations is often limited by the accessible timescales and the computational resources available, posing challenges to comprehensively exploring protein behaviors. Recently emerging approaches have focused on expanding the capability of AlphaFold2 (AF2) to predict conformational substates of protein. Here, we benchmark the performance of various workflows that have adapted AF2 for ensemble prediction and compare the obtained structures with ensembles obtained from MD simulations and NMR. We provide an overview of the levels of performance and accessible timescales that can currently be achieved with machine learning (ML) based ensemble generation. Significant minima of the free energy surfaces remain undetected.
期刊介绍:
Structure aims to publish papers of exceptional interest in the field of structural biology. The journal strives to be essential reading for structural biologists, as well as biologists and biochemists that are interested in macromolecular structure and function. Structure strongly encourages the submission of manuscripts that present structural and molecular insights into biological function and mechanism. Other reports that address fundamental questions in structural biology, such as structure-based examinations of protein evolution, folding, and/or design, will also be considered. We will consider the application of any method, experimental or computational, at high or low resolution, to conduct structural investigations, as long as the method is appropriate for the biological, functional, and mechanistic question(s) being addressed. Likewise, reports describing single-molecule analysis of biological mechanisms are welcome.
In general, the editors encourage submission of experimental structural studies that are enriched by an analysis of structure-activity relationships and will not consider studies that solely report structural information unless the structure or analysis is of exceptional and broad interest. Studies reporting only homology models, de novo models, or molecular dynamics simulations are also discouraged unless the models are informed by or validated by novel experimental data; rationalization of a large body of existing experimental evidence and making testable predictions based on a model or simulation is often not considered sufficient.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
