Wentao Song, Assimo Maris, Charlotte N. Cummings, Luca Evangelisti, Nicholas R. Walker, Sonia Melandri
{"title":"旋转光谱和量子化学计算的强相互作用揭示了半胱胺--H2O 的挑战性构象格局","authors":"Wentao Song, Assimo Maris, Charlotte N. Cummings, Luca Evangelisti, Nicholas R. Walker, Sonia Melandri","doi":"10.1021/acs.jpclett.4c02353","DOIUrl":null,"url":null,"abstract":"A 1:1 molecular complex of cysteamine with water is shown to adopt a cage-like structure where cysteamine accepts a relatively strong hydrogen bond from water while also engaging in two additional weaker interactions (SH···O<sub>w</sub> and CH···O<sub>w</sub>). Experimental and theoretical approaches confirm this conformer as the global minimum on the potential energy surface. Fitting of key structural parameters to experimentally determined moments of inertia yields consistent and accurate results for rotational and <sup>14</sup>N nuclear quadrupole coupling constants which are shown to be challenging to calculate using <i>ab initio</i> methods. Comprehensive analysis of the intermolecular interactions and a thorough comparison with the properties of aminoethanol–water is presented, utilizing independent gradient models based on Hirshfeld partition, quantum theory of atoms-in-molecules, and symmetry-adapted perturbation theory approaches. As expected, the OH group of aminoethanol is a stronger hydrogen bond donor than the SH group in cysteamine, while the CH···O<sub>w</sub> interaction is a key determining factor of the conformational landscape in both cysteamine–water and aminoethanol–water complexes. The results show very clearly that the synergy between theoretical calculations and experimental results is not only desirable but mandatory to get the right answers in such complex conformational surfaces. The results are also clear benchmarks for the accuracy of different theoretical methods in assessing the structures and energy order of the conformations.","PeriodicalId":62,"journal":{"name":"The Journal of Physical Chemistry Letters","volume":"8 1","pages":""},"PeriodicalIF":4.5000,"publicationDate":"2024-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"The Challenging Conformational Landscape of Cysteamine···H2O Revealed by the Strong Interplay of Rotational Spectroscopy and Quantum Chemical Calculations\",\"authors\":\"Wentao Song, Assimo Maris, Charlotte N. Cummings, Luca Evangelisti, Nicholas R. Walker, Sonia Melandri\",\"doi\":\"10.1021/acs.jpclett.4c02353\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"A 1:1 molecular complex of cysteamine with water is shown to adopt a cage-like structure where cysteamine accepts a relatively strong hydrogen bond from water while also engaging in two additional weaker interactions (SH···O<sub>w</sub> and CH···O<sub>w</sub>). Experimental and theoretical approaches confirm this conformer as the global minimum on the potential energy surface. Fitting of key structural parameters to experimentally determined moments of inertia yields consistent and accurate results for rotational and <sup>14</sup>N nuclear quadrupole coupling constants which are shown to be challenging to calculate using <i>ab initio</i> methods. Comprehensive analysis of the intermolecular interactions and a thorough comparison with the properties of aminoethanol–water is presented, utilizing independent gradient models based on Hirshfeld partition, quantum theory of atoms-in-molecules, and symmetry-adapted perturbation theory approaches. As expected, the OH group of aminoethanol is a stronger hydrogen bond donor than the SH group in cysteamine, while the CH···O<sub>w</sub> interaction is a key determining factor of the conformational landscape in both cysteamine–water and aminoethanol–water complexes. The results show very clearly that the synergy between theoretical calculations and experimental results is not only desirable but mandatory to get the right answers in such complex conformational surfaces. The results are also clear benchmarks for the accuracy of different theoretical methods in assessing the structures and energy order of the conformations.\",\"PeriodicalId\":62,\"journal\":{\"name\":\"The Journal of Physical Chemistry Letters\",\"volume\":\"8 1\",\"pages\":\"\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2024-09-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry Letters\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpclett.4c02353\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry Letters","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpclett.4c02353","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
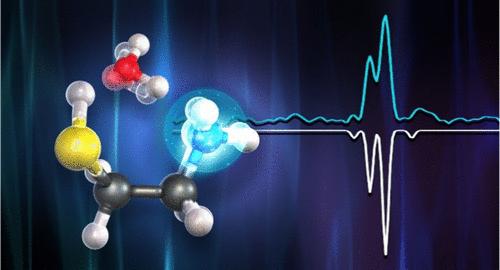
研究表明,半胱胺与水的 1:1 分子复合物采用了笼状结构,其中半胱胺从水中接受了一个相对较强的氢键,同时还参与了另外两个较弱的相互作用(SH--Ow 和 CH--Ow)。实验和理论方法都证实这种构象是势能面上的全局最小值。将关键的结构参数与实验测定的惯性矩进行拟合,得到了一致而准确的旋转和 14N 核四极耦合常数结果,而这些结果表明,使用 ab initio 方法计算这些结果具有挑战性。利用基于 Hirshfeld 分配的独立梯度模型、分子中原子的量子理论和对称性适应扰动理论方法,对分子间相互作用进行了全面分析,并与氨基乙醇-水的性质进行了深入比较。不出所料,氨基乙醇的 OH 基团是比半胱胺的 SH 基团更强的氢键供体,而 CH-Ow 相互作用是半胱胺-水和氨基乙醇-水复合物构象格局的关键决定因素。研究结果清楚地表明,理论计算与实验结果之间的协同作用不仅是可取的,而且是在此类复杂构象表面中获得正确答案的必要条件。这些结果也是不同理论方法评估构象结构和能级的准确性的明确基准。
The Challenging Conformational Landscape of Cysteamine···H2O Revealed by the Strong Interplay of Rotational Spectroscopy and Quantum Chemical Calculations
A 1:1 molecular complex of cysteamine with water is shown to adopt a cage-like structure where cysteamine accepts a relatively strong hydrogen bond from water while also engaging in two additional weaker interactions (SH···Ow and CH···Ow). Experimental and theoretical approaches confirm this conformer as the global minimum on the potential energy surface. Fitting of key structural parameters to experimentally determined moments of inertia yields consistent and accurate results for rotational and 14N nuclear quadrupole coupling constants which are shown to be challenging to calculate using ab initio methods. Comprehensive analysis of the intermolecular interactions and a thorough comparison with the properties of aminoethanol–water is presented, utilizing independent gradient models based on Hirshfeld partition, quantum theory of atoms-in-molecules, and symmetry-adapted perturbation theory approaches. As expected, the OH group of aminoethanol is a stronger hydrogen bond donor than the SH group in cysteamine, while the CH···Ow interaction is a key determining factor of the conformational landscape in both cysteamine–water and aminoethanol–water complexes. The results show very clearly that the synergy between theoretical calculations and experimental results is not only desirable but mandatory to get the right answers in such complex conformational surfaces. The results are also clear benchmarks for the accuracy of different theoretical methods in assessing the structures and energy order of the conformations.
期刊介绍:
The Journal of Physical Chemistry (JPC) Letters is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, chemical physicists, physicists, material scientists, and engineers. An important criterion for acceptance is that the paper reports a significant scientific advance and/or physical insight such that rapid publication is essential. Two issues of JPC Letters are published each month.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
