下载PDF
{"title":"从单细胞开始的外显子组测序","authors":"Ioanna Andreou, Markus Storbeck, Peter Hahn, Samuel Rulli, Eric Lader","doi":"10.1002/cpz1.70017","DOIUrl":null,"url":null,"abstract":"<p>Single-cell genomic analysis enables researchers to gain novel insights across diverse research areas, including developmental biology, tumor heterogeneity, and disease pathogenesis. Conducting single-cell genomic analysis using next-generation sequencing (NGS) methods has traditionally been challenging as the amount of genomic DNA present in a single cell is limited. Advancements in multiple displacement amplification (MDA) technologies allow the unbiased amplification of limited quantities of DNA under conditions that maintain its integrity. This method of amplification results in high yield and facilitates the generation of high-complexity NGS libraries that ensure the highest coverage to effectively allow variant calling.</p><p>With the introduction of new sequencing platforms and chemistry, whole genome sequencing became a more cost-effective application, but enrichment of specific regions of interest further reduces the amount of required sequencing output and associated costs. There are two enrichment methods, polymerase chain reaction (PCR)–based and hybrid-capture-based methods. PCR-based methods are very flexible and highly effective but focus on specific loci, typically known to be associated with disease. Inherited diseases of unknown genetic origin require a more comprehensive approach to capture the genetic variation that is not yet associated with a specific disease. Hybrid capture enrichment methods require considerable amounts of DNA such that exome enrichment from single cells is only possible after preamplification of this limited material.</p><p>This article describes the complete workflow from single cells and small quantities of DNA to exome-NGS libraries for Illumina sequencing instruments and includes the following protocols: © 2024 QIAGEN GmbH. Current Protocols published by Wiley Periodicals LLC.</p><p>This article was corrected on 04 October 2024. See the end of the full text for details.</p><p><b>Basic Protocol 1</b>: Whole genome amplification from single cells or small amounts of gDNA</p><p><b>Basic Protocol 2</b>: NGS library generation of MDA-amplified material</p><p><b>Basic Protocol 3</b>: Exome enrichment</p>","PeriodicalId":93970,"journal":{"name":"Current protocols","volume":"4 9","pages":""},"PeriodicalIF":2.2000,"publicationDate":"2024-09-26","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpz1.70017","citationCount":"0","resultStr":"{\"title\":\"Exome Sequencing Starting from Single Cells\",\"authors\":\"Ioanna Andreou, Markus Storbeck, Peter Hahn, Samuel Rulli, Eric Lader\",\"doi\":\"10.1002/cpz1.70017\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>Single-cell genomic analysis enables researchers to gain novel insights across diverse research areas, including developmental biology, tumor heterogeneity, and disease pathogenesis. Conducting single-cell genomic analysis using next-generation sequencing (NGS) methods has traditionally been challenging as the amount of genomic DNA present in a single cell is limited. Advancements in multiple displacement amplification (MDA) technologies allow the unbiased amplification of limited quantities of DNA under conditions that maintain its integrity. This method of amplification results in high yield and facilitates the generation of high-complexity NGS libraries that ensure the highest coverage to effectively allow variant calling.</p><p>With the introduction of new sequencing platforms and chemistry, whole genome sequencing became a more cost-effective application, but enrichment of specific regions of interest further reduces the amount of required sequencing output and associated costs. There are two enrichment methods, polymerase chain reaction (PCR)–based and hybrid-capture-based methods. PCR-based methods are very flexible and highly effective but focus on specific loci, typically known to be associated with disease. Inherited diseases of unknown genetic origin require a more comprehensive approach to capture the genetic variation that is not yet associated with a specific disease. Hybrid capture enrichment methods require considerable amounts of DNA such that exome enrichment from single cells is only possible after preamplification of this limited material.</p><p>This article describes the complete workflow from single cells and small quantities of DNA to exome-NGS libraries for Illumina sequencing instruments and includes the following protocols: © 2024 QIAGEN GmbH. Current Protocols published by Wiley Periodicals LLC.</p><p>This article was corrected on 04 October 2024. See the end of the full text for details.</p><p><b>Basic Protocol 1</b>: Whole genome amplification from single cells or small amounts of gDNA</p><p><b>Basic Protocol 2</b>: NGS library generation of MDA-amplified material</p><p><b>Basic Protocol 3</b>: Exome enrichment</p>\",\"PeriodicalId\":93970,\"journal\":{\"name\":\"Current protocols\",\"volume\":\"4 9\",\"pages\":\"\"},\"PeriodicalIF\":2.2000,\"publicationDate\":\"2024-09-26\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/cpz1.70017\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Current protocols\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/cpz1.70017\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"\",\"JCRName\":\"\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Current protocols","FirstCategoryId":"1085","ListUrlMain":"https://currentprotocols.onlinelibrary.wiley.com/doi/10.1002/cpz1.70017","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"","JCRName":"","Score":null,"Total":0}
引用次数: 0
引用
批量引用
Exome Sequencing Starting from Single Cells
Single-cell genomic analysis enables researchers to gain novel insights across diverse research areas, including developmental biology, tumor heterogeneity, and disease pathogenesis. Conducting single-cell genomic analysis using next-generation sequencing (NGS) methods has traditionally been challenging as the amount of genomic DNA present in a single cell is limited. Advancements in multiple displacement amplification (MDA) technologies allow the unbiased amplification of limited quantities of DNA under conditions that maintain its integrity. This method of amplification results in high yield and facilitates the generation of high-complexity NGS libraries that ensure the highest coverage to effectively allow variant calling.
With the introduction of new sequencing platforms and chemistry, whole genome sequencing became a more cost-effective application, but enrichment of specific regions of interest further reduces the amount of required sequencing output and associated costs. There are two enrichment methods, polymerase chain reaction (PCR)–based and hybrid-capture-based methods. PCR-based methods are very flexible and highly effective but focus on specific loci, typically known to be associated with disease. Inherited diseases of unknown genetic origin require a more comprehensive approach to capture the genetic variation that is not yet associated with a specific disease. Hybrid capture enrichment methods require considerable amounts of DNA such that exome enrichment from single cells is only possible after preamplification of this limited material.
This article describes the complete workflow from single cells and small quantities of DNA to exome-NGS libraries for Illumina sequencing instruments and includes the following protocols: © 2024 QIAGEN GmbH. Current Protocols published by Wiley Periodicals LLC.
This article was corrected on 04 October 2024. See the end of the full text for details.
Basic Protocol 1 : Whole genome amplification from single cells or small amounts of gDNA
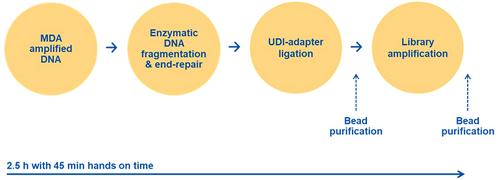
Basic Protocol 2 : NGS library generation of MDA-amplified material
Basic Protocol 3 : Exome enrichment




 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
