Jesi Lee, Dean Joseph Tantillo, Lee-Ping Wang, Oliver Fiehn
{"title":"利用 Ab Initio 分子动力学预测碰撞诱导解离串联质谱 (CID-MS/MS)","authors":"Jesi Lee, Dean Joseph Tantillo, Lee-Ping Wang, Oliver Fiehn","doi":"10.1021/acs.jcim.4c00760","DOIUrl":null,"url":null,"abstract":"Compound identification is at the center of metabolomics, usually by comparing experimental mass spectra against library spectra. However, most compounds are not commercially available to generate library spectra. Hence, for such compounds, MS/MS spectra need to be predicted. Machine learning and heuristic models have largely failed except for lipids. Here, quantum chemistry software can be used to predict mass spectra. However, quantum chemistry predictions for collision induced dissociation (CID) mass spectra in LC-MS/MS are rare. We present the CIDMD (Collision-Induced Dissociation via Molecular Dynamics) framework to model CID-based MS/MS spectra. It uses first-principles molecular dynamics (MD) to simulate the physical process of molecular collisions in CID tandem mass spectrometry. First, molecular ions are constructed at specific protonation sites. Using density functional theory, these protonated ions are targeted by argon collider gas atoms at user-specified velocities. Subsequent bond breakages are simulated over time for at least 1,000 fs. Each simulation is repeated multiple times from various collisional directions. Fragmentations are accumulated over those repeated collisions to generate CIDMD in silico mass spectra. Twelve small metabolites (<205 Da) were selected to test the accuracy of this framework in comparison to experimental MS/MS spectra. When testing different protomers, collider velocities, number of simulations, simulation time and impact factor b cutoffs, we yielded 261 predicted mass spectra. These in silico spectra resulted in entropy similarity scores of an average 624 ± 189 for all 261 spectra compared to their corresponding experimental spectra, which improved to 828 ± 77 when using optimal parameters of the most probable protomers for 12 molecules. With increasing molecular mass, higher velocities achieved better results. Similarly, different protomers showed large differences in fragmentation; hence, with increasing numbers of protomers and tautomers, the average CIDMD prediction accuracy decreased. Mechanistic details showed that specific fragment ions can be produced from different protomers via multiple fragmentation pathways. We propose that CIDMD is a suitable tool to predict mass spectra of small metabolites like produced by the gut microbiome.","PeriodicalId":44,"journal":{"name":"Journal of Chemical Information and Modeling ","volume":"6 1","pages":""},"PeriodicalIF":6.4000,"publicationDate":"2024-09-27","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Predicting Collision-Induced-Dissociation Tandem Mass Spectra (CID-MS/MS) Using Ab Initio Molecular Dynamics\",\"authors\":\"Jesi Lee, Dean Joseph Tantillo, Lee-Ping Wang, Oliver Fiehn\",\"doi\":\"10.1021/acs.jcim.4c00760\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Compound identification is at the center of metabolomics, usually by comparing experimental mass spectra against library spectra. However, most compounds are not commercially available to generate library spectra. Hence, for such compounds, MS/MS spectra need to be predicted. Machine learning and heuristic models have largely failed except for lipids. Here, quantum chemistry software can be used to predict mass spectra. However, quantum chemistry predictions for collision induced dissociation (CID) mass spectra in LC-MS/MS are rare. We present the CIDMD (Collision-Induced Dissociation via Molecular Dynamics) framework to model CID-based MS/MS spectra. It uses first-principles molecular dynamics (MD) to simulate the physical process of molecular collisions in CID tandem mass spectrometry. First, molecular ions are constructed at specific protonation sites. Using density functional theory, these protonated ions are targeted by argon collider gas atoms at user-specified velocities. Subsequent bond breakages are simulated over time for at least 1,000 fs. Each simulation is repeated multiple times from various collisional directions. Fragmentations are accumulated over those repeated collisions to generate CIDMD in silico mass spectra. Twelve small metabolites (<205 Da) were selected to test the accuracy of this framework in comparison to experimental MS/MS spectra. When testing different protomers, collider velocities, number of simulations, simulation time and impact factor b cutoffs, we yielded 261 predicted mass spectra. These in silico spectra resulted in entropy similarity scores of an average 624 ± 189 for all 261 spectra compared to their corresponding experimental spectra, which improved to 828 ± 77 when using optimal parameters of the most probable protomers for 12 molecules. With increasing molecular mass, higher velocities achieved better results. Similarly, different protomers showed large differences in fragmentation; hence, with increasing numbers of protomers and tautomers, the average CIDMD prediction accuracy decreased. Mechanistic details showed that specific fragment ions can be produced from different protomers via multiple fragmentation pathways. We propose that CIDMD is a suitable tool to predict mass spectra of small metabolites like produced by the gut microbiome.\",\"PeriodicalId\":44,\"journal\":{\"name\":\"Journal of Chemical Information and Modeling \",\"volume\":\"6 1\",\"pages\":\"\"},\"PeriodicalIF\":6.4000,\"publicationDate\":\"2024-09-27\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Information and Modeling \",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jcim.4c00760\",\"RegionNum\":2,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"CHEMISTRY, MEDICINAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Information and Modeling ","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jcim.4c00760","RegionNum":2,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"CHEMISTRY, MEDICINAL","Score":null,"Total":0}
Predicting Collision-Induced-Dissociation Tandem Mass Spectra (CID-MS/MS) Using Ab Initio Molecular Dynamics
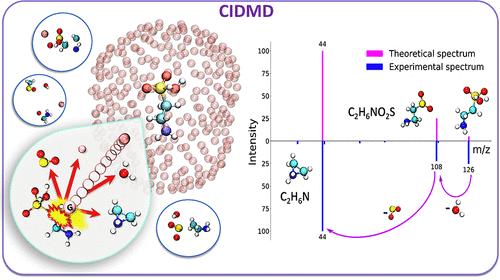
Compound identification is at the center of metabolomics, usually by comparing experimental mass spectra against library spectra. However, most compounds are not commercially available to generate library spectra. Hence, for such compounds, MS/MS spectra need to be predicted. Machine learning and heuristic models have largely failed except for lipids. Here, quantum chemistry software can be used to predict mass spectra. However, quantum chemistry predictions for collision induced dissociation (CID) mass spectra in LC-MS/MS are rare. We present the CIDMD (Collision-Induced Dissociation via Molecular Dynamics) framework to model CID-based MS/MS spectra. It uses first-principles molecular dynamics (MD) to simulate the physical process of molecular collisions in CID tandem mass spectrometry. First, molecular ions are constructed at specific protonation sites. Using density functional theory, these protonated ions are targeted by argon collider gas atoms at user-specified velocities. Subsequent bond breakages are simulated over time for at least 1,000 fs. Each simulation is repeated multiple times from various collisional directions. Fragmentations are accumulated over those repeated collisions to generate CIDMD in silico mass spectra. Twelve small metabolites (<205 Da) were selected to test the accuracy of this framework in comparison to experimental MS/MS spectra. When testing different protomers, collider velocities, number of simulations, simulation time and impact factor b cutoffs, we yielded 261 predicted mass spectra. These in silico spectra resulted in entropy similarity scores of an average 624 ± 189 for all 261 spectra compared to their corresponding experimental spectra, which improved to 828 ± 77 when using optimal parameters of the most probable protomers for 12 molecules. With increasing molecular mass, higher velocities achieved better results. Similarly, different protomers showed large differences in fragmentation; hence, with increasing numbers of protomers and tautomers, the average CIDMD prediction accuracy decreased. Mechanistic details showed that specific fragment ions can be produced from different protomers via multiple fragmentation pathways. We propose that CIDMD is a suitable tool to predict mass spectra of small metabolites like produced by the gut microbiome.
期刊介绍:
The Journal of Chemical Information and Modeling publishes papers reporting new methodology and/or important applications in the fields of chemical informatics and molecular modeling. Specific topics include the representation and computer-based searching of chemical databases, molecular modeling, computer-aided molecular design of new materials, catalysts, or ligands, development of new computational methods or efficient algorithms for chemical software, and biopharmaceutical chemistry including analyses of biological activity and other issues related to drug discovery.
Astute chemists, computer scientists, and information specialists look to this monthly’s insightful research studies, programming innovations, and software reviews to keep current with advances in this integral, multidisciplinary field.
As a subscriber you’ll stay abreast of database search systems, use of graph theory in chemical problems, substructure search systems, pattern recognition and clustering, analysis of chemical and physical data, molecular modeling, graphics and natural language interfaces, bibliometric and citation analysis, and synthesis design and reactions databases.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
