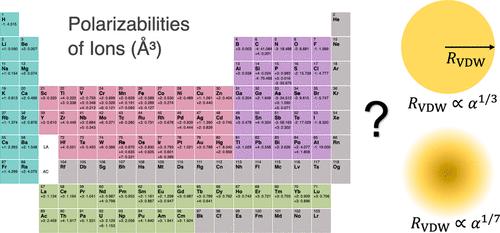
{"title":"探索离子极化性及其与范德华半径的相关性:理论研究","authors":"Madelyn Smith, Richa Khatiwada, Pengfei Li","doi":"10.1021/acs.jctc.4c00632","DOIUrl":null,"url":null,"abstract":"Polarizability (α) is a fundamental property which measures the tendency of the electron cloud of an atom, ion, or molecule to be distorted by electric field. Polarizability contributes to important physical properties such as molecular interactions or dielectric constants; thus, it is essential to have accurate polarizabilities in molecular simulations. However, it remains a challenge to develop polarizable force fields (FFs) for ions in computational chemistry. In particular, a comprehensive set of polarizabilities for ions has not been derived. Herein, we derived a systematic set of polarizabilities for atoms and ions across the periodic table based on high-level quantum mechanics calculations. These values have excellent agreement with experimental data. Furthermore, we examined the relationship between the obtained polarizabilities and the van der Waals (VDW) radii (<i>R</i><sub>VDW</sub>) that we previously determined (<i>J. Chem. Theory Comput.</i>, <b>2023</b>, <i>19</i>, 2064). Two relationships, <i>R</i><sub>VDW</sub> ∝ α<sup>1/7</sup> and <i>R</i><sub>VDW</sub> ∝ α<sup>1/3</sup>, proposed in previous studies were examined in the present work. Our results indicated the former relationship, which was derived based on the quantum harmonic oscillator model, prevails for atoms and cations, but neither relationship provides a satisfactory fit for anions. This is consistent with the tight-binding nature of the electrons in atoms and cations, while it is more challenging to quantify the polarizabilities of anions because of their more dispersed electron clouds. Moreover, we compared different approaches to determine the dispersion coefficients, including the London equation, Slater–Kirkwood equation, symmetry-adapted perturbation theory (SAPT) calculations, and time-dependent density functional theory method, along with the approach based on VDW constants. Our results indicated that although different approaches predict deviated magnitudes for the dispersion coefficients, their predictions are highly correlated, implying that each of these approaches can be used to evaluate dispersion interactions after proper scaling. Finally, we have developed a parametrization strategy for the 12-6-4 model based on the obtained insights. We specifically compared the performance of the 12-6-4 model with SAPT and SobEDA analyses to model interactions involving Na<sup>+</sup>/Mg<sup>2+</sup> and various ligands containing He, Ne, Ar, H<sub>2</sub>O, NH<sub>3</sub>, [H<sub>2</sub>PO<sub>4</sub>]<sup>−</sup>, and [HPO<sub>4</sub>]<sup>2–</sup>. Our results demonstrate that the 12-6-4 parameters effectively reproduce both the total interaction energy and the individual energy components (electrostatics, exchange-repulsion, dispersion, and induction), highlighting the physical robustness of the 12-6-4 model and the effectiveness of our parametrization approach. This study has significant implications for advancing the development of next-generation ion models and polarizable FFs.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"22 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Exploring Ion Polarizabilities and Their Correlation with van der Waals Radii: A Theoretical Investigation\",\"authors\":\"Madelyn Smith, Richa Khatiwada, Pengfei Li\",\"doi\":\"10.1021/acs.jctc.4c00632\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Polarizability (α) is a fundamental property which measures the tendency of the electron cloud of an atom, ion, or molecule to be distorted by electric field. Polarizability contributes to important physical properties such as molecular interactions or dielectric constants; thus, it is essential to have accurate polarizabilities in molecular simulations. However, it remains a challenge to develop polarizable force fields (FFs) for ions in computational chemistry. In particular, a comprehensive set of polarizabilities for ions has not been derived. Herein, we derived a systematic set of polarizabilities for atoms and ions across the periodic table based on high-level quantum mechanics calculations. These values have excellent agreement with experimental data. Furthermore, we examined the relationship between the obtained polarizabilities and the van der Waals (VDW) radii (<i>R</i><sub>VDW</sub>) that we previously determined (<i>J. Chem. Theory Comput.</i>, <b>2023</b>, <i>19</i>, 2064). Two relationships, <i>R</i><sub>VDW</sub> ∝ α<sup>1/7</sup> and <i>R</i><sub>VDW</sub> ∝ α<sup>1/3</sup>, proposed in previous studies were examined in the present work. Our results indicated the former relationship, which was derived based on the quantum harmonic oscillator model, prevails for atoms and cations, but neither relationship provides a satisfactory fit for anions. This is consistent with the tight-binding nature of the electrons in atoms and cations, while it is more challenging to quantify the polarizabilities of anions because of their more dispersed electron clouds. Moreover, we compared different approaches to determine the dispersion coefficients, including the London equation, Slater–Kirkwood equation, symmetry-adapted perturbation theory (SAPT) calculations, and time-dependent density functional theory method, along with the approach based on VDW constants. Our results indicated that although different approaches predict deviated magnitudes for the dispersion coefficients, their predictions are highly correlated, implying that each of these approaches can be used to evaluate dispersion interactions after proper scaling. Finally, we have developed a parametrization strategy for the 12-6-4 model based on the obtained insights. We specifically compared the performance of the 12-6-4 model with SAPT and SobEDA analyses to model interactions involving Na<sup>+</sup>/Mg<sup>2+</sup> and various ligands containing He, Ne, Ar, H<sub>2</sub>O, NH<sub>3</sub>, [H<sub>2</sub>PO<sub>4</sub>]<sup>−</sup>, and [HPO<sub>4</sub>]<sup>2–</sup>. Our results demonstrate that the 12-6-4 parameters effectively reproduce both the total interaction energy and the individual energy components (electrostatics, exchange-repulsion, dispersion, and induction), highlighting the physical robustness of the 12-6-4 model and the effectiveness of our parametrization approach. This study has significant implications for advancing the development of next-generation ion models and polarizable FFs.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"22 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00632\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00632","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Exploring Ion Polarizabilities and Their Correlation with van der Waals Radii: A Theoretical Investigation
Polarizability (α) is a fundamental property which measures the tendency of the electron cloud of an atom, ion, or molecule to be distorted by electric field. Polarizability contributes to important physical properties such as molecular interactions or dielectric constants; thus, it is essential to have accurate polarizabilities in molecular simulations. However, it remains a challenge to develop polarizable force fields (FFs) for ions in computational chemistry. In particular, a comprehensive set of polarizabilities for ions has not been derived. Herein, we derived a systematic set of polarizabilities for atoms and ions across the periodic table based on high-level quantum mechanics calculations. These values have excellent agreement with experimental data. Furthermore, we examined the relationship between the obtained polarizabilities and the van der Waals (VDW) radii (RVDW) that we previously determined (J. Chem. Theory Comput., 2023, 19, 2064). Two relationships, RVDW ∝ α1/7 and RVDW ∝ α1/3, proposed in previous studies were examined in the present work. Our results indicated the former relationship, which was derived based on the quantum harmonic oscillator model, prevails for atoms and cations, but neither relationship provides a satisfactory fit for anions. This is consistent with the tight-binding nature of the electrons in atoms and cations, while it is more challenging to quantify the polarizabilities of anions because of their more dispersed electron clouds. Moreover, we compared different approaches to determine the dispersion coefficients, including the London equation, Slater–Kirkwood equation, symmetry-adapted perturbation theory (SAPT) calculations, and time-dependent density functional theory method, along with the approach based on VDW constants. Our results indicated that although different approaches predict deviated magnitudes for the dispersion coefficients, their predictions are highly correlated, implying that each of these approaches can be used to evaluate dispersion interactions after proper scaling. Finally, we have developed a parametrization strategy for the 12-6-4 model based on the obtained insights. We specifically compared the performance of the 12-6-4 model with SAPT and SobEDA analyses to model interactions involving Na+/Mg2+ and various ligands containing He, Ne, Ar, H2O, NH3, [H2PO4]−, and [HPO4]2–. Our results demonstrate that the 12-6-4 parameters effectively reproduce both the total interaction energy and the individual energy components (electrostatics, exchange-repulsion, dispersion, and induction), highlighting the physical robustness of the 12-6-4 model and the effectiveness of our parametrization approach. This study has significant implications for advancing the development of next-generation ion models and polarizable FFs.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
