{"title":"半导体带隙的蒙特卡洛显式相关二阶多体格林函数计算","authors":"J. César Cruz, So Hirata","doi":"10.1021/acs.jpcc.4c03848","DOIUrl":null,"url":null,"abstract":"A systematically converging series of <i>ab initio</i>, postdensity functional, size-consistent, electron-correlated approximations is desired for predictive computing of electronic band structures of insulating, semiconducting, and metallic solids. A series that meets all of these desiderata (except the applicability to metals) is <i>ab initio</i> many-body Green’s function theory based on Gaussian-type orbital (GTO) basis sets. Here, its leading-order approximation, the second-order Green’s function (GF2) method in the diagonal and frequency-independent approximations with the aug-cc-pVDZ basis set, is applied to the fundamental band gaps of three semiconductors (diamond, silicon, and silicon carbide in the zincblende structure) using cluster models. Corrections are made to the basis set incompleteness errors by the explicit correlation (F12) ansatz (GF2-F12) for the valence band edges. The crystals are modeled as surface-passivated clusters of increasing sizes, whose wave functions are expanded by up to 2709 GTO basis functions. Immense computational costs of these calculations are overcome by the highly scalable stochastic algorithm of the Monte Carlo GF2-F12 method, whose operation cost per state increases only as a cubic power of system size, which has a tiny memory footprint and easily achieves near-perfect parallel efficiency on thousands of CPUs or on hundreds of GPUs. The correlated, F12-corrected highest-occupied and lowest-unoccupied molecular orbital energy (HOMO–LUMO) gap is 5.78 ± 0.07 eV for C<sub>87</sub>H<sub>76</sub> as compared with the experimental value of the fundamental (indirect) band gap of bulk diamond at 5.48 eV. The correlated, F12-corrected HOMO–LUMO gaps for Si<sub>75</sub>H<sub>76</sub> and Si<sub>32</sub>C<sub>43</sub>H<sub>76</sub> are 2.56 ± 0.15 and 3.50 ± 0.12 eV, respectively, which are expected to decrease further with increasing cluster sizes. The experimental fundamental (indirect) band gaps of bulk silicon and silicon carbide are 1.17 and 2.42 eV, respectively.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"3 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Monte Carlo Explicitly Correlated Second-Order Many-Body Green’s Function Calculations of Semiconductor Band Gaps\",\"authors\":\"J. César Cruz, So Hirata\",\"doi\":\"10.1021/acs.jpcc.4c03848\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"A systematically converging series of <i>ab initio</i>, postdensity functional, size-consistent, electron-correlated approximations is desired for predictive computing of electronic band structures of insulating, semiconducting, and metallic solids. A series that meets all of these desiderata (except the applicability to metals) is <i>ab initio</i> many-body Green’s function theory based on Gaussian-type orbital (GTO) basis sets. Here, its leading-order approximation, the second-order Green’s function (GF2) method in the diagonal and frequency-independent approximations with the aug-cc-pVDZ basis set, is applied to the fundamental band gaps of three semiconductors (diamond, silicon, and silicon carbide in the zincblende structure) using cluster models. Corrections are made to the basis set incompleteness errors by the explicit correlation (F12) ansatz (GF2-F12) for the valence band edges. The crystals are modeled as surface-passivated clusters of increasing sizes, whose wave functions are expanded by up to 2709 GTO basis functions. Immense computational costs of these calculations are overcome by the highly scalable stochastic algorithm of the Monte Carlo GF2-F12 method, whose operation cost per state increases only as a cubic power of system size, which has a tiny memory footprint and easily achieves near-perfect parallel efficiency on thousands of CPUs or on hundreds of GPUs. The correlated, F12-corrected highest-occupied and lowest-unoccupied molecular orbital energy (HOMO–LUMO) gap is 5.78 ± 0.07 eV for C<sub>87</sub>H<sub>76</sub> as compared with the experimental value of the fundamental (indirect) band gap of bulk diamond at 5.48 eV. The correlated, F12-corrected HOMO–LUMO gaps for Si<sub>75</sub>H<sub>76</sub> and Si<sub>32</sub>C<sub>43</sub>H<sub>76</sub> are 2.56 ± 0.15 and 3.50 ± 0.12 eV, respectively, which are expected to decrease further with increasing cluster sizes. The experimental fundamental (indirect) band gaps of bulk silicon and silicon carbide are 1.17 and 2.42 eV, respectively.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"3 1\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.4c03848\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c03848","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Monte Carlo Explicitly Correlated Second-Order Many-Body Green’s Function Calculations of Semiconductor Band Gaps
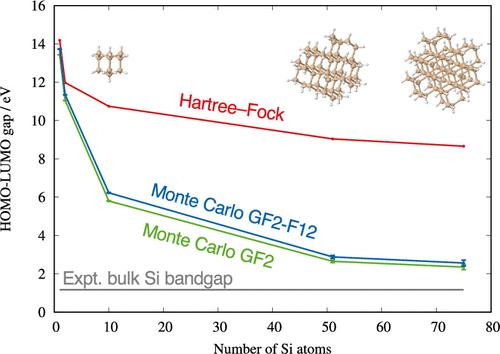
A systematically converging series of ab initio, postdensity functional, size-consistent, electron-correlated approximations is desired for predictive computing of electronic band structures of insulating, semiconducting, and metallic solids. A series that meets all of these desiderata (except the applicability to metals) is ab initio many-body Green’s function theory based on Gaussian-type orbital (GTO) basis sets. Here, its leading-order approximation, the second-order Green’s function (GF2) method in the diagonal and frequency-independent approximations with the aug-cc-pVDZ basis set, is applied to the fundamental band gaps of three semiconductors (diamond, silicon, and silicon carbide in the zincblende structure) using cluster models. Corrections are made to the basis set incompleteness errors by the explicit correlation (F12) ansatz (GF2-F12) for the valence band edges. The crystals are modeled as surface-passivated clusters of increasing sizes, whose wave functions are expanded by up to 2709 GTO basis functions. Immense computational costs of these calculations are overcome by the highly scalable stochastic algorithm of the Monte Carlo GF2-F12 method, whose operation cost per state increases only as a cubic power of system size, which has a tiny memory footprint and easily achieves near-perfect parallel efficiency on thousands of CPUs or on hundreds of GPUs. The correlated, F12-corrected highest-occupied and lowest-unoccupied molecular orbital energy (HOMO–LUMO) gap is 5.78 ± 0.07 eV for C87H76 as compared with the experimental value of the fundamental (indirect) band gap of bulk diamond at 5.48 eV. The correlated, F12-corrected HOMO–LUMO gaps for Si75H76 and Si32C43H76 are 2.56 ± 0.15 and 3.50 ± 0.12 eV, respectively, which are expected to decrease further with increasing cluster sizes. The experimental fundamental (indirect) band gaps of bulk silicon and silicon carbide are 1.17 and 2.42 eV, respectively.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
