{"title":"温度和压力对 2,6-二氨基-3,5-二硝基吡嗪-1-氧化物晶体分解机制的影响:ab initio 分子动力学研究。","authors":"Jincheng Ji, Hui Li, Weihua Zhu","doi":"10.1007/s00894-024-06156-z","DOIUrl":null,"url":null,"abstract":"<div><h3>Context</h3><p>The decomposition process of 2,6-diamino-3,5-dinitropyrazine-1-oxide (LLM-105) crystal at high temperatures (2500 and 3390 K) and detonation pressure of 33.4 GPa coupled with temperatures were studied by ab initio molecular dynamics simulations. The results show that the initial decomposition mechanism of LLM-105 is the same under different conditions. The product analysis indicates that high temperature is conducive to the formation of N<sub>2</sub> and CO<sub>2</sub>, but inhibited the formation of H<sub>2</sub>O. It is found that the formation mechanism of H<sub>2</sub>O is the same under different conditions, which involves the reaction between OH radical and H radical. Although the detailed processes of the formation of N<sub>2</sub> are different, they all involve the reaction between nitrogen-containing fragments, and its core is the formation of intermediates with R<sub>1</sub>-NN-R<sub>2</sub> structure. The core of the formation of CO<sub>2</sub> under different conditions is to form the intermediate R<sub>1</sub>-CO-R<sub>2</sub> with carbonyl structure, and then generate the fragment with -OCO- structure, and finally generate CO<sub>2</sub>. This research may provide new insights into the initiation and subsequent decomposition mechanisms of energetic materials under extreme conditions.</p><h3>Methods</h3><p>The LLM-105 supercell was constructed using the Materials Studio 7.0 package. AIMD simulations were performed in the CASTEP package. AIMD simulations adopted NVT and NPT ensemble, and the temperature was controlled by Nosé thermostat, while the pressure was controlled by Andersen barostat. Besides, DFT calculations were carried out at the B3LYP/6–311 + G(d,p) level using the Gaussian 09 package.</p></div>","PeriodicalId":651,"journal":{"name":"Journal of Molecular Modeling","volume":"30 10","pages":""},"PeriodicalIF":2.5000,"publicationDate":"2024-09-28","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Temperature and pressure effects on the decomposition mechanisms of 2,6-diamino-3,5-dinitropyrazine-1-oxide crystal: ab initio molecular dynamics study\",\"authors\":\"Jincheng Ji, Hui Li, Weihua Zhu\",\"doi\":\"10.1007/s00894-024-06156-z\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Context</h3><p>The decomposition process of 2,6-diamino-3,5-dinitropyrazine-1-oxide (LLM-105) crystal at high temperatures (2500 and 3390 K) and detonation pressure of 33.4 GPa coupled with temperatures were studied by ab initio molecular dynamics simulations. The results show that the initial decomposition mechanism of LLM-105 is the same under different conditions. The product analysis indicates that high temperature is conducive to the formation of N<sub>2</sub> and CO<sub>2</sub>, but inhibited the formation of H<sub>2</sub>O. It is found that the formation mechanism of H<sub>2</sub>O is the same under different conditions, which involves the reaction between OH radical and H radical. Although the detailed processes of the formation of N<sub>2</sub> are different, they all involve the reaction between nitrogen-containing fragments, and its core is the formation of intermediates with R<sub>1</sub>-NN-R<sub>2</sub> structure. The core of the formation of CO<sub>2</sub> under different conditions is to form the intermediate R<sub>1</sub>-CO-R<sub>2</sub> with carbonyl structure, and then generate the fragment with -OCO- structure, and finally generate CO<sub>2</sub>. This research may provide new insights into the initiation and subsequent decomposition mechanisms of energetic materials under extreme conditions.</p><h3>Methods</h3><p>The LLM-105 supercell was constructed using the Materials Studio 7.0 package. AIMD simulations were performed in the CASTEP package. AIMD simulations adopted NVT and NPT ensemble, and the temperature was controlled by Nosé thermostat, while the pressure was controlled by Andersen barostat. Besides, DFT calculations were carried out at the B3LYP/6–311 + G(d,p) level using the Gaussian 09 package.</p></div>\",\"PeriodicalId\":651,\"journal\":{\"name\":\"Journal of Molecular Modeling\",\"volume\":\"30 10\",\"pages\":\"\"},\"PeriodicalIF\":2.5000,\"publicationDate\":\"2024-09-28\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Modeling\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://link.springer.com/article/10.1007/s00894-024-06156-z\",\"RegionNum\":4,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Modeling","FirstCategoryId":"92","ListUrlMain":"https://link.springer.com/article/10.1007/s00894-024-06156-z","RegionNum":4,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
Temperature and pressure effects on the decomposition mechanisms of 2,6-diamino-3,5-dinitropyrazine-1-oxide crystal: ab initio molecular dynamics study
Context
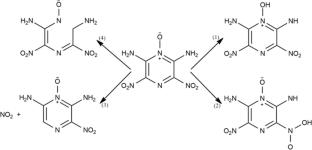
The decomposition process of 2,6-diamino-3,5-dinitropyrazine-1-oxide (LLM-105) crystal at high temperatures (2500 and 3390 K) and detonation pressure of 33.4 GPa coupled with temperatures were studied by ab initio molecular dynamics simulations. The results show that the initial decomposition mechanism of LLM-105 is the same under different conditions. The product analysis indicates that high temperature is conducive to the formation of N2 and CO2, but inhibited the formation of H2O. It is found that the formation mechanism of H2O is the same under different conditions, which involves the reaction between OH radical and H radical. Although the detailed processes of the formation of N2 are different, they all involve the reaction between nitrogen-containing fragments, and its core is the formation of intermediates with R1-NN-R2 structure. The core of the formation of CO2 under different conditions is to form the intermediate R1-CO-R2 with carbonyl structure, and then generate the fragment with -OCO- structure, and finally generate CO2. This research may provide new insights into the initiation and subsequent decomposition mechanisms of energetic materials under extreme conditions.
Methods
The LLM-105 supercell was constructed using the Materials Studio 7.0 package. AIMD simulations were performed in the CASTEP package. AIMD simulations adopted NVT and NPT ensemble, and the temperature was controlled by Nosé thermostat, while the pressure was controlled by Andersen barostat. Besides, DFT calculations were carried out at the B3LYP/6–311 + G(d,p) level using the Gaussian 09 package.
期刊介绍:
The Journal of Molecular Modeling focuses on "hardcore" modeling, publishing high-quality research and reports. Founded in 1995 as a purely electronic journal, it has adapted its format to include a full-color print edition, and adjusted its aims and scope fit the fast-changing field of molecular modeling, with a particular focus on three-dimensional modeling.
Today, the journal covers all aspects of molecular modeling including life science modeling; materials modeling; new methods; and computational chemistry.
Topics include computer-aided molecular design; rational drug design, de novo ligand design, receptor modeling and docking; cheminformatics, data analysis, visualization and mining; computational medicinal chemistry; homology modeling; simulation of peptides, DNA and other biopolymers; quantitative structure-activity relationships (QSAR) and ADME-modeling; modeling of biological reaction mechanisms; and combined experimental and computational studies in which calculations play a major role.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
