Vinod K Viswanathan, Aloke G Ghoshal, Anant Mohan, Ketaki Patil, Chaitanya Bhargave, Sanjay Choudhari, Suyog Mehta
{"title":"在特发性肺纤维化患者中使用 Nintedanib 进行基于患者特征的管理","authors":"Vinod K Viswanathan, Aloke G Ghoshal, Anant Mohan, Ketaki Patil, Chaitanya Bhargave, Sanjay Choudhari, Suyog Mehta","doi":"10.1007/s41030-024-00271-1","DOIUrl":null,"url":null,"abstract":"<p><p>A severe and progressive interstitial lung disease (ILD) known as idiopathic pulmonary fibrosis (IPF) has an unknown etiology with poorly defined mechanisms of development. Among the currently prescribed pharmacological interventions for IPF, nintedanib demonstrates the ability to decelerate the deterioration of lung function and yield positive clinical outcomes. Multiple randomized placebo-controlled trials have confirmed the efficacy and acceptable safety profile of nintedanib. Real-world evidence studies also support the use of nintedanib in IPF, being an efficient and well-tolerated treatment option. It has the potential to stabilize the disease progression in patients with ILD. Patients with IPF frequently have comorbidities like diabetes and hypertension, which can exacerbate the course of disease, reduce quality of life, and decrease treatment adherence. For well-informed decision-making, it is important for healthcare professionals to recognize the position of nintedanib therapy in IPF with comorbidities. The gastrointestinal adverse effects, notably diarrhea, dominate the nintedanib safety profile. These can be effectively controlled by closely monitoring side effects, administering anti-diarrheal and anti-emetic drugs, reducing the nintedanib dose, and discontinuing it in case of severe symptoms with an option to reintroduce the treatment after side effects subside. Symptomatic interventions and monitoring of liver enzymes may reduce the occurrence of permanent treatment discontinuations.</p>","PeriodicalId":20919,"journal":{"name":"Pulmonary Therapy","volume":" ","pages":"377-409"},"PeriodicalIF":3.0000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11573957/pdf/","citationCount":"0","resultStr":"{\"title\":\"Patient Profile-Based Management with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis.\",\"authors\":\"Vinod K Viswanathan, Aloke G Ghoshal, Anant Mohan, Ketaki Patil, Chaitanya Bhargave, Sanjay Choudhari, Suyog Mehta\",\"doi\":\"10.1007/s41030-024-00271-1\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>A severe and progressive interstitial lung disease (ILD) known as idiopathic pulmonary fibrosis (IPF) has an unknown etiology with poorly defined mechanisms of development. Among the currently prescribed pharmacological interventions for IPF, nintedanib demonstrates the ability to decelerate the deterioration of lung function and yield positive clinical outcomes. Multiple randomized placebo-controlled trials have confirmed the efficacy and acceptable safety profile of nintedanib. Real-world evidence studies also support the use of nintedanib in IPF, being an efficient and well-tolerated treatment option. It has the potential to stabilize the disease progression in patients with ILD. Patients with IPF frequently have comorbidities like diabetes and hypertension, which can exacerbate the course of disease, reduce quality of life, and decrease treatment adherence. For well-informed decision-making, it is important for healthcare professionals to recognize the position of nintedanib therapy in IPF with comorbidities. The gastrointestinal adverse effects, notably diarrhea, dominate the nintedanib safety profile. These can be effectively controlled by closely monitoring side effects, administering anti-diarrheal and anti-emetic drugs, reducing the nintedanib dose, and discontinuing it in case of severe symptoms with an option to reintroduce the treatment after side effects subside. Symptomatic interventions and monitoring of liver enzymes may reduce the occurrence of permanent treatment discontinuations.</p>\",\"PeriodicalId\":20919,\"journal\":{\"name\":\"Pulmonary Therapy\",\"volume\":\" \",\"pages\":\"377-409\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11573957/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Pulmonary Therapy\",\"FirstCategoryId\":\"1085\",\"ListUrlMain\":\"https://doi.org/10.1007/s41030-024-00271-1\",\"RegionNum\":0,\"RegionCategory\":null,\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/28 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q2\",\"JCRName\":\"RESPIRATORY SYSTEM\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Pulmonary Therapy","FirstCategoryId":"1085","ListUrlMain":"https://doi.org/10.1007/s41030-024-00271-1","RegionNum":0,"RegionCategory":null,"ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/28 0:00:00","PubModel":"Epub","JCR":"Q2","JCRName":"RESPIRATORY SYSTEM","Score":null,"Total":0}
Patient Profile-Based Management with Nintedanib in Patients with Idiopathic Pulmonary Fibrosis.
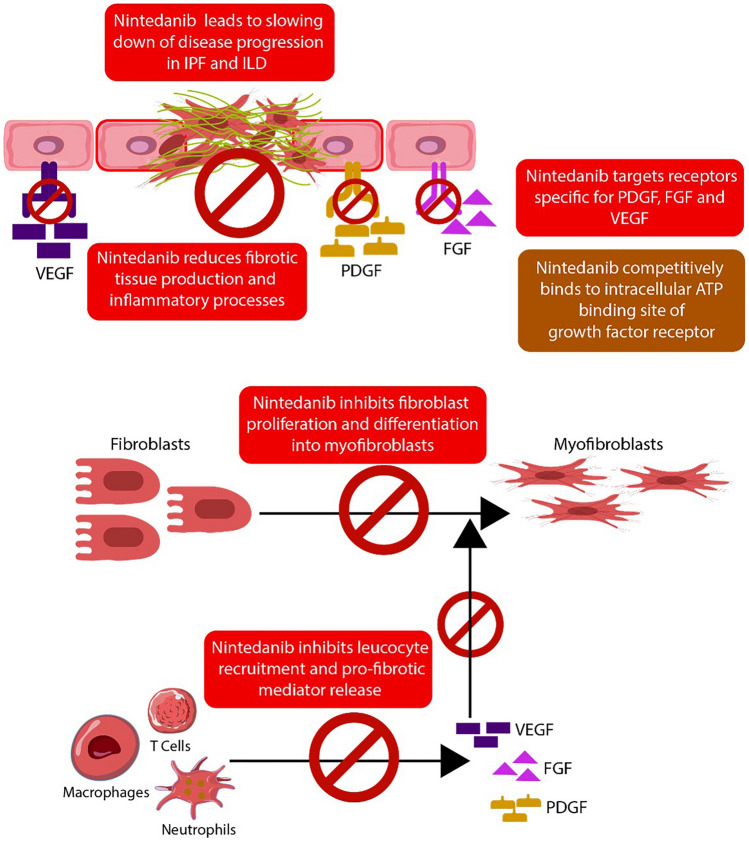
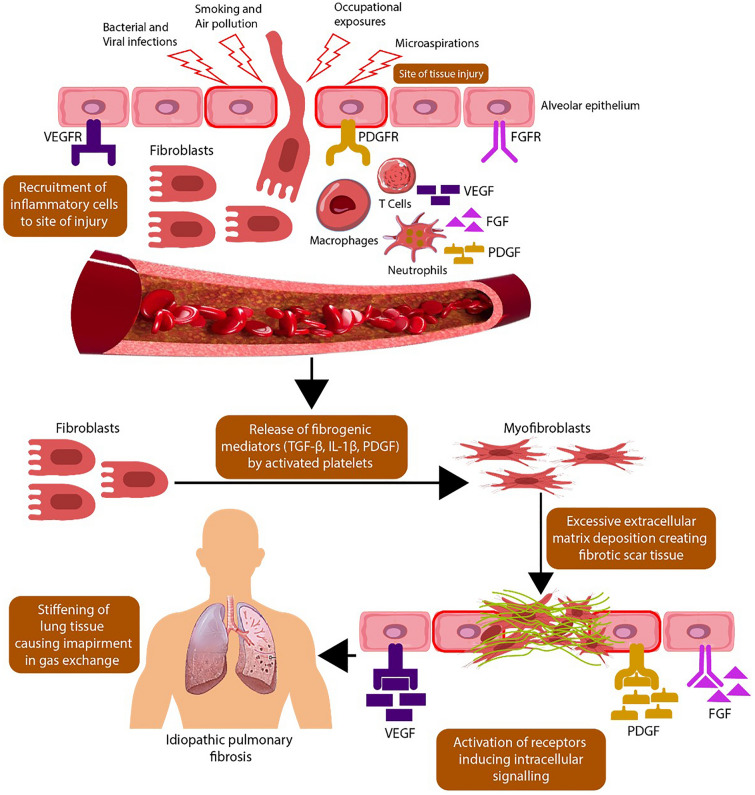
A severe and progressive interstitial lung disease (ILD) known as idiopathic pulmonary fibrosis (IPF) has an unknown etiology with poorly defined mechanisms of development. Among the currently prescribed pharmacological interventions for IPF, nintedanib demonstrates the ability to decelerate the deterioration of lung function and yield positive clinical outcomes. Multiple randomized placebo-controlled trials have confirmed the efficacy and acceptable safety profile of nintedanib. Real-world evidence studies also support the use of nintedanib in IPF, being an efficient and well-tolerated treatment option. It has the potential to stabilize the disease progression in patients with ILD. Patients with IPF frequently have comorbidities like diabetes and hypertension, which can exacerbate the course of disease, reduce quality of life, and decrease treatment adherence. For well-informed decision-making, it is important for healthcare professionals to recognize the position of nintedanib therapy in IPF with comorbidities. The gastrointestinal adverse effects, notably diarrhea, dominate the nintedanib safety profile. These can be effectively controlled by closely monitoring side effects, administering anti-diarrheal and anti-emetic drugs, reducing the nintedanib dose, and discontinuing it in case of severe symptoms with an option to reintroduce the treatment after side effects subside. Symptomatic interventions and monitoring of liver enzymes may reduce the occurrence of permanent treatment discontinuations.
期刊介绍:
Aims and Scope
Pulmonary Therapy is an international, open access, peer-reviewed (single-blind), and rapid publication journal. The scope of the journal is broad and will consider all scientifically sound research from pre-clinical, clinical (all phases), observational, real-world, and health outcomes research around the use of pulmonary therapies, devices, and surgical techniques.
Areas of focus include, but are not limited to: asthma; chronic obstructive pulmonary disease; idiopathic pulmonary fibrosis; pulmonary hypertension; cystic fibrosis; lung cancer; respiratory tract disorders; allergic rhinitis and other respiratory allergies; influenza, pneumococcal infection, respiratory syncytial virus and other respiratory infections; and inhalers and other device therapies.
The journal is of interest to a broad audience of pharmaceutical and healthcare professionals and publishes original research, reviews, case reports/series, trial protocols and short communications such as commentaries and editorials. Pulmonary Therapy will consider all scientifically sound research be it positive, confirmatory or negative data. Submissions are welcomed whether they relate to an international and/or a country-specific audience, something that is crucially important when researchers are trying to target more specific patient populations. This inclusive approach allows the journal to assist in the dissemination of quality research, which may be considered of insufficient interest by other journals.
Rapid Publication
The journal’s publication timelines aim for a rapid peer review of 2 weeks. If an article is accepted it will be published 3–4 weeks from acceptance. The rapid timelines are achieved through the combination of a dedicated in-house editorial team, who manage article workflow, and an extensive Editorial and Advisory Board who assist with peer review. This allows the journal to support the rapid dissemination of research, whilst still providing robust peer review. Combined with the journal’s open access model this allows for the rapid, efficient communication of the latest research and reviews, fostering the advancement of pulmonary therapies.
Open Access
All articles published by Pulmonary Therapy are open access.
Personal Service
The journal’s dedicated in-house editorial team offer a personal “concierge service” meaning authors will always have an editorial contact able to update them on the status of their manuscript. The editorial team check all manuscripts to ensure that articles conform to the most recent COPE, GPP and ICMJE publishing guidelines. This supports the publication of ethically sound and transparent research.
Digital Features and Plain Language Summaries
Pulmonary Therapy offers a range of additional features designed to increase the visibility, readership and educational value of the journal’s content. Each article is accompanied by key summary points, giving a time-efficient overview of the content to a wide readership. Articles may be accompanied by plain language summaries to assist readers who have some knowledge of, but not in-depth expertise in, the area to understand the scientific content and overall implications of the article. The journal also provides the option to include various types of digital features including animated abstracts, video abstracts, slide decks, audio slides, instructional videos, infographics, podcasts and animations. All additional features are peer reviewed to the same high standard as the article itself. If you consider that your paper would benefit from the inclusion of a digital feature, please let us know. Our editorial team are able to create high-quality slide decks and infographics in-house, and video abstracts through our partner Research Square, and would be happy to assist in any way we can. For further information about digital features, please contact the journal editor (see ‘Contact the Journal’ for email address), and see the ‘Guidelines for digital features and plain language summaries’ document under ‘Submission guidelines’.
For examples of digital features please visit our showcase page https://springerhealthcare.com/expertise/publishing-digital-features/
Publication Fees
Upon acceptance of an article, authors will be required to pay the mandatory Rapid Service Fee of €4500/ $5100/ £3650. The journal will consider fee discounts and waivers for developing countries and this is decided on a case by case basis.
Peer Review Process
Upon submission, manuscripts are assessed by the editorial team to ensure they fit within the aims and scope of the journal and are also checked for plagiarism. All suitable submissions are then subject to a comprehensive single-blind peer review. Reviewers are selected based on their relevant expertise and publication history in the subject area. The journal has an extensive pool of editorial and advisory board members who have been selected to assist with peer review based on the afore-mentioned criteria.
At least two extensive reviews are required to make the editorial decision, with the exception of some article types such as Commentaries, Editorials, and Letters which are generally reviewed by one member of the Editorial Board. Where reviewer recommendations are conflicted, the editorial board will be contacted for further advice and a presiding decision. Manuscripts are then either accepted, rejected or authors are required to make major or minor revisions (both reviewer comments and editorial comments may need to be addressed). Once a revised manuscript is re-submitted, it is assessed along with the responses to reviewer comments and if it has been adequately revised it will be accepted for publication. Accepted manuscripts are then copyedited and typeset by the production team before online publication. Appeals against decisions following peer review are considered on a case-by-case basis and should be sent to the journal editor.
Preprints
We encourage posting of preprints of primary research manuscripts on preprint servers, authors’ or institutional websites, and open communications between researchers whether on community preprint servers or preprint commenting platforms. Posting of preprints is not considered prior publication and will not jeopardize consideration in our journals. Authors should disclose details of preprint posting during the submission process or at any other point during consideration in one of our journals. Once the manuscript is published, it is the author’s responsibility to ensure that the preprint record is updated with a publication reference, including the DOI and a URL link to the published version of the article on the journal website.
Please follow the link for further information on preprint sharing:
https://www.springer.com/gp/authors-editors/journal-author/journal-author-helpdesk/submission/1302#c16721550
Copyright
Pulmonary Therapy''s content is published open access under the Creative Commons Attribution-Noncommercial License, which allows users to read, copy, distribute, and make derivative works for non-commercial purposes from the material, as long as the author of the original work is cited. The author assigns the exclusive right to any commercial use of the article to Springer. For more information about the Creative Commons Attribution-Noncommercial License, click here: http://creativecommons.org/licenses/by-nc/4.0.
Contact
For more information about the journal, including pre-submission enquiries, please contact christopher.vautrinot@springer.com.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
