Javier Poyatos-García, Patricia Soblechero-Martín, Alessandro Liquori, Andrea López-Martínez, Pilar Maestre, Elisa González-Romero, Rafael P Vázquez-Manrique, Nuria Muelas, Gema García-García, Jessica Ohana, Virginia Arechavala-Gomeza, Juan J Vílchez
{"title":"DMD 基因第 45 至 55 号外显子的缺失:从治疗角度到体外模型。","authors":"Javier Poyatos-García, Patricia Soblechero-Martín, Alessandro Liquori, Andrea López-Martínez, Pilar Maestre, Elisa González-Romero, Rafael P Vázquez-Manrique, Nuria Muelas, Gema García-García, Jessica Ohana, Virginia Arechavala-Gomeza, Juan J Vílchez","doi":"10.1186/s13395-024-00353-3","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Gene editing therapies in development for correcting out-of-frame DMD mutations in Duchenne muscular dystrophy aim to replicate benign spontaneous deletions. Deletion of 45-55 DMD exons (del45-55) was described in asymptomatic subjects, but recently serious skeletal and cardiac complications have been reported. Uncovering why a single mutation like del45-55 is able to induce diverse phenotypes and grades of severity may impact the strategies of emerging therapies. Cellular models are essential for this purpose, but their availability is compromised by scarce muscle biopsies.</p><p><strong>Methods: </strong>We introduced, as a proof-of-concept, using CRISPR-Cas9 edition, a del45-55 mimicking the intronic breakpoints harboured by a subset of patients of this form of dystrophinopathy (designing specific gRNAs), into a Duchenne patient's cell line. The edited cell line was characterized evaluating the dystrophin expression and the myogenic status.</p><p><strong>Results: </strong>Dystrophin expression was restored, and the myogenic defects were ameliorated in the edited myoblasts harbouring a specific del45-55. Besides confirming the potential of CRISPR-Cas9 to create tailored mutations (despite the low cleavage efficiency of our gRNAs) as a useful approach to generate in vitro models, we also generated an immortalized myoblast line derived from a patient with a specific del45-55.</p><p><strong>Conclusions: </strong>Overall, we provide helpful resources to deepen into unknown factors responsible for DMD-pathophysiology.</p>","PeriodicalId":21747,"journal":{"name":"Skeletal Muscle","volume":"14 1","pages":"21"},"PeriodicalIF":3.9000,"publicationDate":"2024-10-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11443720/pdf/","citationCount":"0","resultStr":"{\"title\":\"Deletion of exons 45 to 55 in the DMD gene: from the therapeutic perspective to the in vitro model.\",\"authors\":\"Javier Poyatos-García, Patricia Soblechero-Martín, Alessandro Liquori, Andrea López-Martínez, Pilar Maestre, Elisa González-Romero, Rafael P Vázquez-Manrique, Nuria Muelas, Gema García-García, Jessica Ohana, Virginia Arechavala-Gomeza, Juan J Vílchez\",\"doi\":\"10.1186/s13395-024-00353-3\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Gene editing therapies in development for correcting out-of-frame DMD mutations in Duchenne muscular dystrophy aim to replicate benign spontaneous deletions. Deletion of 45-55 DMD exons (del45-55) was described in asymptomatic subjects, but recently serious skeletal and cardiac complications have been reported. Uncovering why a single mutation like del45-55 is able to induce diverse phenotypes and grades of severity may impact the strategies of emerging therapies. Cellular models are essential for this purpose, but their availability is compromised by scarce muscle biopsies.</p><p><strong>Methods: </strong>We introduced, as a proof-of-concept, using CRISPR-Cas9 edition, a del45-55 mimicking the intronic breakpoints harboured by a subset of patients of this form of dystrophinopathy (designing specific gRNAs), into a Duchenne patient's cell line. The edited cell line was characterized evaluating the dystrophin expression and the myogenic status.</p><p><strong>Results: </strong>Dystrophin expression was restored, and the myogenic defects were ameliorated in the edited myoblasts harbouring a specific del45-55. Besides confirming the potential of CRISPR-Cas9 to create tailored mutations (despite the low cleavage efficiency of our gRNAs) as a useful approach to generate in vitro models, we also generated an immortalized myoblast line derived from a patient with a specific del45-55.</p><p><strong>Conclusions: </strong>Overall, we provide helpful resources to deepen into unknown factors responsible for DMD-pathophysiology.</p>\",\"PeriodicalId\":21747,\"journal\":{\"name\":\"Skeletal Muscle\",\"volume\":\"14 1\",\"pages\":\"21\"},\"PeriodicalIF\":3.9000,\"publicationDate\":\"2024-10-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11443720/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Skeletal Muscle\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1186/s13395-024-00353-3\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CELL BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Skeletal Muscle","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1186/s13395-024-00353-3","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CELL BIOLOGY","Score":null,"Total":0}
Deletion of exons 45 to 55 in the DMD gene: from the therapeutic perspective to the in vitro model.
Background: Gene editing therapies in development for correcting out-of-frame DMD mutations in Duchenne muscular dystrophy aim to replicate benign spontaneous deletions. Deletion of 45-55 DMD exons (del45-55) was described in asymptomatic subjects, but recently serious skeletal and cardiac complications have been reported. Uncovering why a single mutation like del45-55 is able to induce diverse phenotypes and grades of severity may impact the strategies of emerging therapies. Cellular models are essential for this purpose, but their availability is compromised by scarce muscle biopsies.
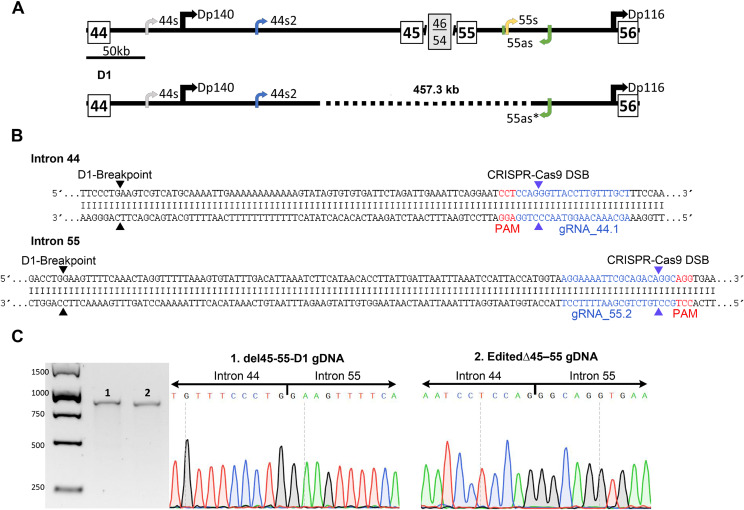
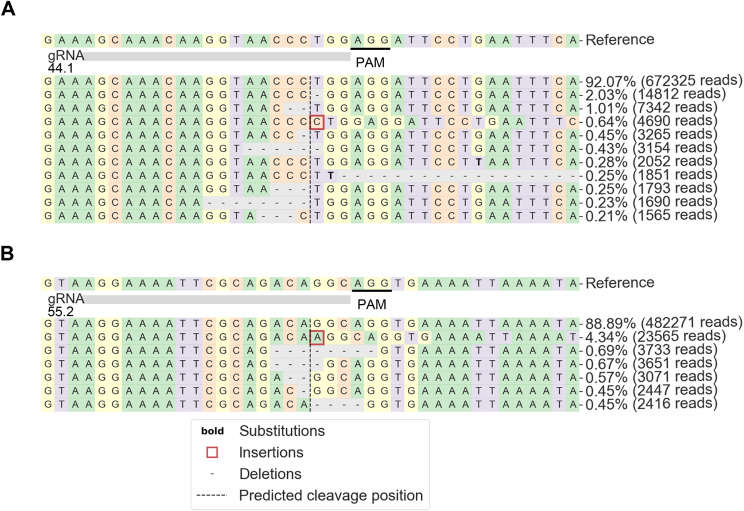
Methods: We introduced, as a proof-of-concept, using CRISPR-Cas9 edition, a del45-55 mimicking the intronic breakpoints harboured by a subset of patients of this form of dystrophinopathy (designing specific gRNAs), into a Duchenne patient's cell line. The edited cell line was characterized evaluating the dystrophin expression and the myogenic status.
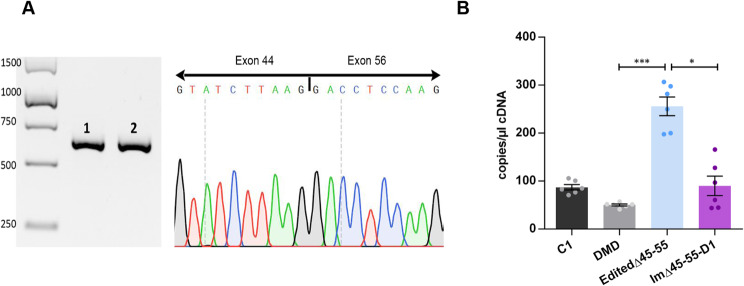
Results: Dystrophin expression was restored, and the myogenic defects were ameliorated in the edited myoblasts harbouring a specific del45-55. Besides confirming the potential of CRISPR-Cas9 to create tailored mutations (despite the low cleavage efficiency of our gRNAs) as a useful approach to generate in vitro models, we also generated an immortalized myoblast line derived from a patient with a specific del45-55.
Conclusions: Overall, we provide helpful resources to deepen into unknown factors responsible for DMD-pathophysiology.
期刊介绍:
The only open access journal in its field, Skeletal Muscle publishes novel, cutting-edge research and technological advancements that investigate the molecular mechanisms underlying the biology of skeletal muscle. Reflecting the breadth of research in this area, the journal welcomes manuscripts about the development, metabolism, the regulation of mass and function, aging, degeneration, dystrophy and regeneration of skeletal muscle, with an emphasis on understanding adult skeletal muscle, its maintenance, and its interactions with non-muscle cell types and regulatory modulators.
Main areas of interest include:
-differentiation of skeletal muscle-
atrophy and hypertrophy of skeletal muscle-
aging of skeletal muscle-
regeneration and degeneration of skeletal muscle-
biology of satellite and satellite-like cells-
dystrophic degeneration of skeletal muscle-
energy and glucose homeostasis in skeletal muscle-
non-dystrophic genetic diseases of skeletal muscle, such as Spinal Muscular Atrophy and myopathies-
maintenance of neuromuscular junctions-
roles of ryanodine receptors and calcium signaling in skeletal muscle-
roles of nuclear receptors in skeletal muscle-
roles of GPCRs and GPCR signaling in skeletal muscle-
other relevant aspects of skeletal muscle biology.
In addition, articles on translational clinical studies that address molecular and cellular mechanisms of skeletal muscle will be published. Case reports are also encouraged for submission.
Skeletal Muscle reflects the breadth of research on skeletal muscle and bridges gaps between diverse areas of science for example cardiac cell biology and neurobiology, which share common features with respect to cell differentiation, excitatory membranes, cell-cell communication, and maintenance. Suitable articles are model and mechanism-driven, and apply statistical principles where appropriate; purely descriptive studies are of lesser interest.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
