{"title":"启用 ANI-2 的开源协议,可在对接后估算配体应变","authors":"Francois Berenger, Koji Tsuda","doi":"10.1002/jcc.27478","DOIUrl":null,"url":null,"abstract":"<p>In protein-ligand docking, the score assigned to a protein-ligand complex is approximate. Especially, the internal energy of the ligand is difficult to compute precisely using a molecular mechanics based force-field, introducing significant noise in the rank-ordering of ligands. We propose an open-source protocol (https://github.com/UnixJunkie/MMO), using two quantum mechanics (QM) single point energy calculations, plus a Monte Carlo (Monte Carlo) based ligand minimization procedure in-between, to estimate ligand strain after docking. The MC simulation uses the ANI-2x (QM approximating) force field and is performed in the dihedral space. On some protein targets, using strain filtering after docking allows to significantly improve hit rates. We performed a structure-based virtual screening campaign on nine protein targets from the Laboratoire d'Innovation Thérapeutique—PubChem assays dataset using Cambridge crystallographic data centre genetic optimization for ligand docking. Then, docked ligands were submitted to the strain estimation protocol and the impact on hit rate was analyzed. As for docking, the method does not always work. However, if sufficient active and inactive molecules are known for a given protein target, its efficiency can be evaluated.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-10-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27478","citationCount":"0","resultStr":"{\"title\":\"An ANI-2 enabled open-source protocol to estimate ligand strain after docking\",\"authors\":\"Francois Berenger, Koji Tsuda\",\"doi\":\"10.1002/jcc.27478\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>In protein-ligand docking, the score assigned to a protein-ligand complex is approximate. Especially, the internal energy of the ligand is difficult to compute precisely using a molecular mechanics based force-field, introducing significant noise in the rank-ordering of ligands. We propose an open-source protocol (https://github.com/UnixJunkie/MMO), using two quantum mechanics (QM) single point energy calculations, plus a Monte Carlo (Monte Carlo) based ligand minimization procedure in-between, to estimate ligand strain after docking. The MC simulation uses the ANI-2x (QM approximating) force field and is performed in the dihedral space. On some protein targets, using strain filtering after docking allows to significantly improve hit rates. We performed a structure-based virtual screening campaign on nine protein targets from the Laboratoire d'Innovation Thérapeutique—PubChem assays dataset using Cambridge crystallographic data centre genetic optimization for ligand docking. Then, docked ligands were submitted to the strain estimation protocol and the impact on hit rate was analyzed. As for docking, the method does not always work. However, if sufficient active and inactive molecules are known for a given protein target, its efficiency can be evaluated.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-10-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://onlinelibrary.wiley.com/doi/epdf/10.1002/jcc.27478\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27478\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27478","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
An ANI-2 enabled open-source protocol to estimate ligand strain after docking
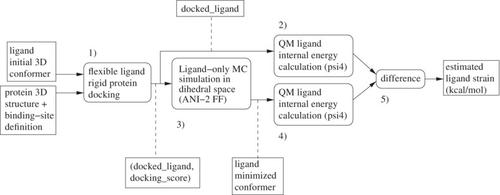
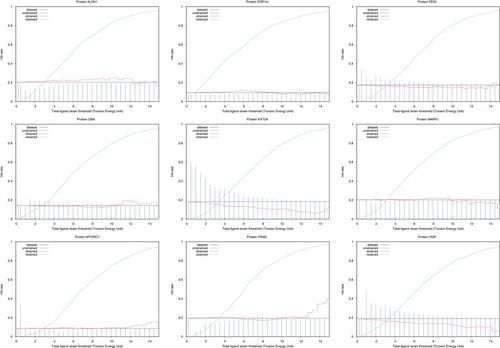
In protein-ligand docking, the score assigned to a protein-ligand complex is approximate. Especially, the internal energy of the ligand is difficult to compute precisely using a molecular mechanics based force-field, introducing significant noise in the rank-ordering of ligands. We propose an open-source protocol (https://github.com/UnixJunkie/MMO), using two quantum mechanics (QM) single point energy calculations, plus a Monte Carlo (Monte Carlo) based ligand minimization procedure in-between, to estimate ligand strain after docking. The MC simulation uses the ANI-2x (QM approximating) force field and is performed in the dihedral space. On some protein targets, using strain filtering after docking allows to significantly improve hit rates. We performed a structure-based virtual screening campaign on nine protein targets from the Laboratoire d'Innovation Thérapeutique—PubChem assays dataset using Cambridge crystallographic data centre genetic optimization for ligand docking. Then, docked ligands were submitted to the strain estimation protocol and the impact on hit rate was analyzed. As for docking, the method does not always work. However, if sufficient active and inactive molecules are known for a given protein target, its efficiency can be evaluated.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
