{"title":"单个氨基酸残基突变导致人类 DNA 连接酶 1 缺乏症(一种罕见的儿科疾病)的机理基础。","authors":"Nikita Zalenski, Yufan He, Zucai Suo","doi":"10.1016/j.jmb.2024.168813","DOIUrl":null,"url":null,"abstract":"<div><div>In mammalian cells, DNA ligase 1 (LIG1) functions as the primary DNA ligase in both genomic replication and single-strand break repair. Several reported mutations in human LIG1, including R305Q, R641L, and R771W, cause LIG1 syndrome, a primary immunodeficiency. While the R641L and R771W mutations, respectively located in the nucleotidyl transferase and oligonucleotide binding domains, have been biochemically characterized and shown to reduce catalytic efficiency, the recently reported R305Q mutation within the DNA binding domain (DBD) remains mechanistically unexplored. The R641L and R771W mutations are known to decrease the catalytic activity of LIG1 by affecting both interdomain interactions and DNA binding during catalysis, without significantly impacting overall DNA affinity. To elucidate the molecular basis of the LIG1 syndrome-causing R305Q mutation, we purified this single-residue mutant protein and investigated its secondary structure, protein stability, DNA binding affinity, and catalytic efficiency. Our findings reveal that the R305Q mutation significantly impairs the function of LIG1 by disrupting the DBD-DNA interactions, leading to a 7–21-fold lower DNA binding affinity and a 33–300-fold reduced catalytic efficiency of LIG1. Additionally, the R305Q mutation slightly decreases LIG1’s protein stability by 2 to 3.6 °C, on par with the effect observed previously with either the R641L or R771W mutant. Collectively, our results uncover a new mechanism whereby the R305Q mutation impairs LIG1-catalyzed nicked DNA ligation, resulting in LIG1 syndrome, and highlight the crucial roles of the DBD-DNA interactions in tight DNA binding and efficient LIG1 catalysis.</div></div>","PeriodicalId":369,"journal":{"name":"Journal of Molecular Biology","volume":"436 22","pages":"Article 168813"},"PeriodicalIF":4.5000,"publicationDate":"2024-11-15","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Mechanistic Basis for a Single Amino Acid Residue Mutation Causing Human DNA Ligase 1 Deficiency, A Rare Pediatric Disease\",\"authors\":\"Nikita Zalenski, Yufan He, Zucai Suo\",\"doi\":\"10.1016/j.jmb.2024.168813\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>In mammalian cells, DNA ligase 1 (LIG1) functions as the primary DNA ligase in both genomic replication and single-strand break repair. Several reported mutations in human LIG1, including R305Q, R641L, and R771W, cause LIG1 syndrome, a primary immunodeficiency. While the R641L and R771W mutations, respectively located in the nucleotidyl transferase and oligonucleotide binding domains, have been biochemically characterized and shown to reduce catalytic efficiency, the recently reported R305Q mutation within the DNA binding domain (DBD) remains mechanistically unexplored. The R641L and R771W mutations are known to decrease the catalytic activity of LIG1 by affecting both interdomain interactions and DNA binding during catalysis, without significantly impacting overall DNA affinity. To elucidate the molecular basis of the LIG1 syndrome-causing R305Q mutation, we purified this single-residue mutant protein and investigated its secondary structure, protein stability, DNA binding affinity, and catalytic efficiency. Our findings reveal that the R305Q mutation significantly impairs the function of LIG1 by disrupting the DBD-DNA interactions, leading to a 7–21-fold lower DNA binding affinity and a 33–300-fold reduced catalytic efficiency of LIG1. Additionally, the R305Q mutation slightly decreases LIG1’s protein stability by 2 to 3.6 °C, on par with the effect observed previously with either the R641L or R771W mutant. Collectively, our results uncover a new mechanism whereby the R305Q mutation impairs LIG1-catalyzed nicked DNA ligation, resulting in LIG1 syndrome, and highlight the crucial roles of the DBD-DNA interactions in tight DNA binding and efficient LIG1 catalysis.</div></div>\",\"PeriodicalId\":369,\"journal\":{\"name\":\"Journal of Molecular Biology\",\"volume\":\"436 22\",\"pages\":\"Article 168813\"},\"PeriodicalIF\":4.5000,\"publicationDate\":\"2024-11-15\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Molecular Biology\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0022283624004352\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/5 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMISTRY & MOLECULAR BIOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Molecular Biology","FirstCategoryId":"99","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0022283624004352","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/5 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"BIOCHEMISTRY & MOLECULAR BIOLOGY","Score":null,"Total":0}
引用次数: 0
摘要
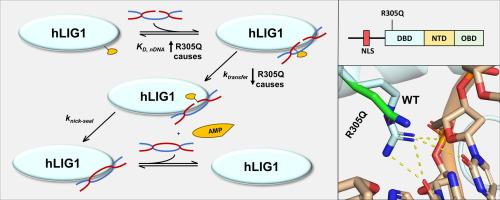
在哺乳动物细胞中,DNA 连接酶 1(LIG1)是基因组复制和单链断裂修复的主要 DNA 连接酶。据报道,人类 LIG1 的几个突变(包括 R305Q、R641L 和 R771W)会导致原发性免疫缺陷 LIG1 综合征。R641L 和 R771W 突变分别位于核苷酸转移酶结构域和寡核苷酸结合结构域,已被生物化学鉴定并证明会降低催化效率,而最近报道的 DNA 结合结构域(DBD)内的 R305Q 突变在机理上仍未得到探讨。众所周知,R641L 和 R771W 突变会在催化过程中影响结构域间的相互作用和 DNA 结合,从而降低 LIG1 的催化活性,但不会显著影响 DNA 的总体亲和力。为了阐明导致 LIG1 综合征的 R305Q 突变的分子基础,我们纯化了这种单残基突变蛋白,并研究了其二级结构、蛋白稳定性、DNA 结合亲和力和催化效率。我们的研究结果表明,R305Q突变通过破坏DBD-DNA相互作用,显著损害了LIG1的功能,导致LIG1的DNA结合亲和力降低了7至21倍,催化效率降低了33至300倍。此外,R305Q 突变会使 LIG1 蛋白的稳定性略微降低 2 到 3.6 °C,与之前观察到的 R641L 或 R771W 突变体的效果相当。总之,我们的研究结果发现了一种新的机制,即 R305Q 突变会损害 LIG1 催化的缺口 DNA 连接,导致 LIG1 综合征,并强调了 DBD-DNA 相互作用在紧密的 DNA 结合和高效的 LIG1 催化中的关键作用。
Mechanistic Basis for a Single Amino Acid Residue Mutation Causing Human DNA Ligase 1 Deficiency, A Rare Pediatric Disease
In mammalian cells, DNA ligase 1 (LIG1) functions as the primary DNA ligase in both genomic replication and single-strand break repair. Several reported mutations in human LIG1, including R305Q, R641L, and R771W, cause LIG1 syndrome, a primary immunodeficiency. While the R641L and R771W mutations, respectively located in the nucleotidyl transferase and oligonucleotide binding domains, have been biochemically characterized and shown to reduce catalytic efficiency, the recently reported R305Q mutation within the DNA binding domain (DBD) remains mechanistically unexplored. The R641L and R771W mutations are known to decrease the catalytic activity of LIG1 by affecting both interdomain interactions and DNA binding during catalysis, without significantly impacting overall DNA affinity. To elucidate the molecular basis of the LIG1 syndrome-causing R305Q mutation, we purified this single-residue mutant protein and investigated its secondary structure, protein stability, DNA binding affinity, and catalytic efficiency. Our findings reveal that the R305Q mutation significantly impairs the function of LIG1 by disrupting the DBD-DNA interactions, leading to a 7–21-fold lower DNA binding affinity and a 33–300-fold reduced catalytic efficiency of LIG1. Additionally, the R305Q mutation slightly decreases LIG1’s protein stability by 2 to 3.6 °C, on par with the effect observed previously with either the R641L or R771W mutant. Collectively, our results uncover a new mechanism whereby the R305Q mutation impairs LIG1-catalyzed nicked DNA ligation, resulting in LIG1 syndrome, and highlight the crucial roles of the DBD-DNA interactions in tight DNA binding and efficient LIG1 catalysis.
期刊介绍:
Journal of Molecular Biology (JMB) provides high quality, comprehensive and broad coverage in all areas of molecular biology. The journal publishes original scientific research papers that provide mechanistic and functional insights and report a significant advance to the field. The journal encourages the submission of multidisciplinary studies that use complementary experimental and computational approaches to address challenging biological questions.
Research areas include but are not limited to: Biomolecular interactions, signaling networks, systems biology; Cell cycle, cell growth, cell differentiation; Cell death, autophagy; Cell signaling and regulation; Chemical biology; Computational biology, in combination with experimental studies; DNA replication, repair, and recombination; Development, regenerative biology, mechanistic and functional studies of stem cells; Epigenetics, chromatin structure and function; Gene expression; Membrane processes, cell surface proteins and cell-cell interactions; Methodological advances, both experimental and theoretical, including databases; Microbiology, virology, and interactions with the host or environment; Microbiota mechanistic and functional studies; Nuclear organization; Post-translational modifications, proteomics; Processing and function of biologically important macromolecules and complexes; Molecular basis of disease; RNA processing, structure and functions of non-coding RNAs, transcription; Sorting, spatiotemporal organization, trafficking; Structural biology; Synthetic biology; Translation, protein folding, chaperones, protein degradation and quality control.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
