Donghu Zhou , Huaduan Zi , Xiaoxi Yang , Xiaojin Li , Yanmeng Li , Anjian Xu , Bei Zhang , Wei Zhang , Xiaojuan Ou , Jidong Jia , Jian Huang , Hong You
{"title":"威尔逊病中 ATP7B 剪接变体因与 COMMD1 的相互作用增强而导致功能障碍。","authors":"Donghu Zhou , Huaduan Zi , Xiaoxi Yang , Xiaojin Li , Yanmeng Li , Anjian Xu , Bei Zhang , Wei Zhang , Xiaojuan Ou , Jidong Jia , Jian Huang , Hong You","doi":"10.1016/j.jcmgh.2024.101418","DOIUrl":null,"url":null,"abstract":"<div><h3>Background & Aims</h3><div>The association between Wilson disease and various <em>ATP7B</em> mutations is well-established; however, the molecular mechanism underlying the functional consequence of these mutations, particularly the splicing mutations, remains unclear. This study focused on the <em>ATP7B</em> c.1543+1G>C variant, to reveal a universal pathogenic mechanism of the ATP7B mutants with altered N-terminus.</div></div><div><h3>Methods</h3><div>The splicing assay and RNA pull-down were performed to explore the mechanism of the aberrant splicing. The <em>ATP7B</em> knockout HuH-7 cell line and <em>Atp7b</em><sup>-/-</sup> mice were created, and the functional consequence of the mutant ATP7B were evaluated <em>in vitro</em> and <em>in vivo</em>.</div></div><div><h3>Results</h3><div>The c.1543+1G>C mutation resulted in the skipping of <em>ATP7B</em> exon 3, and the mutant ATP7B showed a loss of trans-Golgi network localization and was degraded via the ubiquitin-proteasome pathway, facilitated by enhanced interactions with COMMD1. Elevated intercellular copper concentration and reduced survival rate were observed in HuH-7 cells expressing mutant ATP7B. Restoration of wild-type ATP7B in <em>Atp7b</em><sup><em>-/-</em></sup> mice resulted in a substantial improvement in phenotype, whereas mice treated with mutant ATP7B did not demonstrate equivalent benefits.</div></div><div><h3>Conclusion</h3><div>Our research investigated the pathogenicity and mechanism of <em>ATP7B</em> c.1543+1G>C variant, with particular focus on its enhanced interaction with COMMD1 as a potential universal mechanism contributing to the dysfunction of various ATP7B variants. These findings provide a foundation for the development of innovative therapeutic strategies that target abnormal splicing events in a range of hereditary diseases, including Wilson disease.</div></div>","PeriodicalId":55974,"journal":{"name":"Cellular and Molecular Gastroenterology and Hepatology","volume":"19 2","pages":"Article 101418"},"PeriodicalIF":7.1000,"publicationDate":"2024-10-09","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Dysfunction of ATP7B Splicing Variant Caused by Enhanced Interaction With COMMD1 in Wilson Disease\",\"authors\":\"Donghu Zhou , Huaduan Zi , Xiaoxi Yang , Xiaojin Li , Yanmeng Li , Anjian Xu , Bei Zhang , Wei Zhang , Xiaojuan Ou , Jidong Jia , Jian Huang , Hong You\",\"doi\":\"10.1016/j.jcmgh.2024.101418\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background & Aims</h3><div>The association between Wilson disease and various <em>ATP7B</em> mutations is well-established; however, the molecular mechanism underlying the functional consequence of these mutations, particularly the splicing mutations, remains unclear. This study focused on the <em>ATP7B</em> c.1543+1G>C variant, to reveal a universal pathogenic mechanism of the ATP7B mutants with altered N-terminus.</div></div><div><h3>Methods</h3><div>The splicing assay and RNA pull-down were performed to explore the mechanism of the aberrant splicing. The <em>ATP7B</em> knockout HuH-7 cell line and <em>Atp7b</em><sup>-/-</sup> mice were created, and the functional consequence of the mutant ATP7B were evaluated <em>in vitro</em> and <em>in vivo</em>.</div></div><div><h3>Results</h3><div>The c.1543+1G>C mutation resulted in the skipping of <em>ATP7B</em> exon 3, and the mutant ATP7B showed a loss of trans-Golgi network localization and was degraded via the ubiquitin-proteasome pathway, facilitated by enhanced interactions with COMMD1. Elevated intercellular copper concentration and reduced survival rate were observed in HuH-7 cells expressing mutant ATP7B. Restoration of wild-type ATP7B in <em>Atp7b</em><sup><em>-/-</em></sup> mice resulted in a substantial improvement in phenotype, whereas mice treated with mutant ATP7B did not demonstrate equivalent benefits.</div></div><div><h3>Conclusion</h3><div>Our research investigated the pathogenicity and mechanism of <em>ATP7B</em> c.1543+1G>C variant, with particular focus on its enhanced interaction with COMMD1 as a potential universal mechanism contributing to the dysfunction of various ATP7B variants. These findings provide a foundation for the development of innovative therapeutic strategies that target abnormal splicing events in a range of hereditary diseases, including Wilson disease.</div></div>\",\"PeriodicalId\":55974,\"journal\":{\"name\":\"Cellular and Molecular Gastroenterology and Hepatology\",\"volume\":\"19 2\",\"pages\":\"Article 101418\"},\"PeriodicalIF\":7.1000,\"publicationDate\":\"2024-10-09\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Cellular and Molecular Gastroenterology and Hepatology\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2352345X24001735\",\"RegionNum\":1,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GASTROENTEROLOGY & HEPATOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Cellular and Molecular Gastroenterology and Hepatology","FirstCategoryId":"3","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2352345X24001735","RegionNum":1,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GASTROENTEROLOGY & HEPATOLOGY","Score":null,"Total":0}
Dysfunction of ATP7B Splicing Variant Caused by Enhanced Interaction With COMMD1 in Wilson Disease
Background & Aims
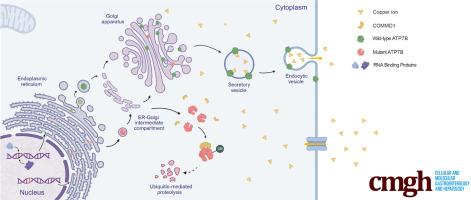
The association between Wilson disease and various ATP7B mutations is well-established; however, the molecular mechanism underlying the functional consequence of these mutations, particularly the splicing mutations, remains unclear. This study focused on the ATP7B c.1543+1G>C variant, to reveal a universal pathogenic mechanism of the ATP7B mutants with altered N-terminus.
Methods
The splicing assay and RNA pull-down were performed to explore the mechanism of the aberrant splicing. The ATP7B knockout HuH-7 cell line and Atp7b-/- mice were created, and the functional consequence of the mutant ATP7B were evaluated in vitro and in vivo.
Results
The c.1543+1G>C mutation resulted in the skipping of ATP7B exon 3, and the mutant ATP7B showed a loss of trans-Golgi network localization and was degraded via the ubiquitin-proteasome pathway, facilitated by enhanced interactions with COMMD1. Elevated intercellular copper concentration and reduced survival rate were observed in HuH-7 cells expressing mutant ATP7B. Restoration of wild-type ATP7B in Atp7b-/- mice resulted in a substantial improvement in phenotype, whereas mice treated with mutant ATP7B did not demonstrate equivalent benefits.
Conclusion
Our research investigated the pathogenicity and mechanism of ATP7B c.1543+1G>C variant, with particular focus on its enhanced interaction with COMMD1 as a potential universal mechanism contributing to the dysfunction of various ATP7B variants. These findings provide a foundation for the development of innovative therapeutic strategies that target abnormal splicing events in a range of hereditary diseases, including Wilson disease.
期刊介绍:
"Cell and Molecular Gastroenterology and Hepatology (CMGH)" is a journal dedicated to advancing the understanding of digestive biology through impactful research that spans the spectrum of normal gastrointestinal, hepatic, and pancreatic functions, as well as their pathologies. The journal's mission is to publish high-quality, hypothesis-driven studies that offer mechanistic novelty and are methodologically robust, covering a wide range of themes in gastroenterology, hepatology, and pancreatology.
CMGH reports on the latest scientific advances in cell biology, immunology, physiology, microbiology, genetics, and neurobiology related to gastrointestinal, hepatobiliary, and pancreatic health and disease. The research published in CMGH is designed to address significant questions in the field, utilizing a variety of experimental approaches, including in vitro models, patient-derived tissues or cells, and animal models. This multifaceted approach enables the journal to contribute to both fundamental discoveries and their translation into clinical applications, ultimately aiming to improve patient care and treatment outcomes in digestive health.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
