Junkai Wang , Yixuan Cai , Shumin Yu , Qianku Hu , Aiguo Zhou , Shaowei Zhang
{"title":"Ti2CO2 MXene 单原子支撑催化剂上甲烷到甲醇转化的第一性原理研究","authors":"Junkai Wang , Yixuan Cai , Shumin Yu , Qianku Hu , Aiguo Zhou , Shaowei Zhang","doi":"10.1016/j.comptc.2024.114892","DOIUrl":null,"url":null,"abstract":"<div><div>Developing efficient catalysts for the conversion of methane (CH<sub>4</sub>) to methanol (CH<sub>3</sub>OH) remains a critical challenge in the chemical industry, with significant implications for both energy production and environmental sustainability. This study pioneers the exploration of the Sc/Ti-Ti<sub>2</sub>CO<sub>2</sub> single-atom catalysts (SACs) for this transformation, utilizing density functional theory (DFT) calculations. Notably, our findings reveal that Sc and Ti are uniquely stable on the Ti<sub>2</sub>CO<sub>2</sub> MXene surface, a discovery that could inform future catalyst designs. We also demonstrate that while CH<sub>4</sub> weakly physisorbs on the Sc/Ti-Ti<sub>2</sub>CO<sub>2</sub> surface, N<sub>2</sub>O molecules decompose directly into N<sub>2</sub> and highly reactive O* species, which bind with Sc/Ti to drive the catalytic process. The oxidation of CH<sub>4</sub> proceeds in two steps: CH<sub>4</sub> + O* → CH<sub>3</sub>* + OH* with reaction barriers of 0.58 eV (Sc) and 1.38 eV (Ti), followed by CH<sub>3</sub>* + OH* → CH<sub>3</sub>OH with barriers of 1.5 eV (Sc) and 1.61 eV (Ti). Importantly, the low desorption energy of CH<sub>3</sub>OH, especially on Sc (0.85 eV), highlights the exceptional catalytic potential of Sc/Ti<sub>2</sub>CO<sub>2</sub> for the direct conversion of CH<sub>4</sub> to CH<sub>3</sub>OH. These results not only underscore the feasibility of using MXene-based SACs for CH<sub>4</sub> oxidation but also provide a theoretical foundation for the development of highly efficient catalysts in this domain.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114892"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"First-principles investigation of methane to methanol conversion on Ti2CO2 MXene supported single-atom catalyst\",\"authors\":\"Junkai Wang , Yixuan Cai , Shumin Yu , Qianku Hu , Aiguo Zhou , Shaowei Zhang\",\"doi\":\"10.1016/j.comptc.2024.114892\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Developing efficient catalysts for the conversion of methane (CH<sub>4</sub>) to methanol (CH<sub>3</sub>OH) remains a critical challenge in the chemical industry, with significant implications for both energy production and environmental sustainability. This study pioneers the exploration of the Sc/Ti-Ti<sub>2</sub>CO<sub>2</sub> single-atom catalysts (SACs) for this transformation, utilizing density functional theory (DFT) calculations. Notably, our findings reveal that Sc and Ti are uniquely stable on the Ti<sub>2</sub>CO<sub>2</sub> MXene surface, a discovery that could inform future catalyst designs. We also demonstrate that while CH<sub>4</sub> weakly physisorbs on the Sc/Ti-Ti<sub>2</sub>CO<sub>2</sub> surface, N<sub>2</sub>O molecules decompose directly into N<sub>2</sub> and highly reactive O* species, which bind with Sc/Ti to drive the catalytic process. The oxidation of CH<sub>4</sub> proceeds in two steps: CH<sub>4</sub> + O* → CH<sub>3</sub>* + OH* with reaction barriers of 0.58 eV (Sc) and 1.38 eV (Ti), followed by CH<sub>3</sub>* + OH* → CH<sub>3</sub>OH with barriers of 1.5 eV (Sc) and 1.61 eV (Ti). Importantly, the low desorption energy of CH<sub>3</sub>OH, especially on Sc (0.85 eV), highlights the exceptional catalytic potential of Sc/Ti<sub>2</sub>CO<sub>2</sub> for the direct conversion of CH<sub>4</sub> to CH<sub>3</sub>OH. These results not only underscore the feasibility of using MXene-based SACs for CH<sub>4</sub> oxidation but also provide a theoretical foundation for the development of highly efficient catalysts in this domain.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1241 \",\"pages\":\"Article 114892\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004316\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004316","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/27 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
First-principles investigation of methane to methanol conversion on Ti2CO2 MXene supported single-atom catalyst
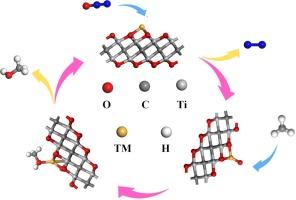
Developing efficient catalysts for the conversion of methane (CH4) to methanol (CH3OH) remains a critical challenge in the chemical industry, with significant implications for both energy production and environmental sustainability. This study pioneers the exploration of the Sc/Ti-Ti2CO2 single-atom catalysts (SACs) for this transformation, utilizing density functional theory (DFT) calculations. Notably, our findings reveal that Sc and Ti are uniquely stable on the Ti2CO2 MXene surface, a discovery that could inform future catalyst designs. We also demonstrate that while CH4 weakly physisorbs on the Sc/Ti-Ti2CO2 surface, N2O molecules decompose directly into N2 and highly reactive O* species, which bind with Sc/Ti to drive the catalytic process. The oxidation of CH4 proceeds in two steps: CH4 + O* → CH3* + OH* with reaction barriers of 0.58 eV (Sc) and 1.38 eV (Ti), followed by CH3* + OH* → CH3OH with barriers of 1.5 eV (Sc) and 1.61 eV (Ti). Importantly, the low desorption energy of CH3OH, especially on Sc (0.85 eV), highlights the exceptional catalytic potential of Sc/Ti2CO2 for the direct conversion of CH4 to CH3OH. These results not only underscore the feasibility of using MXene-based SACs for CH4 oxidation but also provide a theoretical foundation for the development of highly efficient catalysts in this domain.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
