Rahma El Mouhi , Ahmed Slimi , Souad El Khattabi , Adil Touimi Benjelloun , Asmae Fitri , Mohammed Benzakour , Mohammed Mcharfi , Mustafa Kurban
{"title":"用于体异质结太阳能电池 (BHJ) 的基于三苯胺的新型小分子 D-A 的计算研究","authors":"Rahma El Mouhi , Ahmed Slimi , Souad El Khattabi , Adil Touimi Benjelloun , Asmae Fitri , Mohammed Benzakour , Mohammed Mcharfi , Mustafa Kurban","doi":"10.1016/j.comptc.2024.114899","DOIUrl":null,"url":null,"abstract":"<div><div>In this computational study, four donor–acceptor (D–A) small molecules were constructed using indacenodithiophene triphenylamine (IDTTPA) as the donor, with modifications made to the acceptor units intended for use in organic bulk heterojunction (BHJ) solar cells. Their electrical and optical properties were determined through density functional theory (DFT) and time-dependent DFT (TD-DFT) techniques. To ascertain the impact of acceptor group modifications on intramolecular electron delocalization and light absorption capabilities, several key variables were examined. Based on the results, it was determined that molecule SM4 demonstrated the best performance among the designed compounds. It exhibited a maximum wavelength of 594 nm, a narrow energy gap of 1.91 eV, a low-lying HOMO energy level of −4.512 eV in its absorption spectra, and a theoretical power conversion efficiency (PCE) of 8 %. This research provides valuable insights for the development of efficient D-A compounds for use in organic solar cells.</div></div>","PeriodicalId":284,"journal":{"name":"Computational and Theoretical Chemistry","volume":"1241 ","pages":"Article 114899"},"PeriodicalIF":3.0000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Computational study of new small molecules D-A based on triphenylamines for bulk heterojunction solar cells (BHJ)\",\"authors\":\"Rahma El Mouhi , Ahmed Slimi , Souad El Khattabi , Adil Touimi Benjelloun , Asmae Fitri , Mohammed Benzakour , Mohammed Mcharfi , Mustafa Kurban\",\"doi\":\"10.1016/j.comptc.2024.114899\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>In this computational study, four donor–acceptor (D–A) small molecules were constructed using indacenodithiophene triphenylamine (IDTTPA) as the donor, with modifications made to the acceptor units intended for use in organic bulk heterojunction (BHJ) solar cells. Their electrical and optical properties were determined through density functional theory (DFT) and time-dependent DFT (TD-DFT) techniques. To ascertain the impact of acceptor group modifications on intramolecular electron delocalization and light absorption capabilities, several key variables were examined. Based on the results, it was determined that molecule SM4 demonstrated the best performance among the designed compounds. It exhibited a maximum wavelength of 594 nm, a narrow energy gap of 1.91 eV, a low-lying HOMO energy level of −4.512 eV in its absorption spectra, and a theoretical power conversion efficiency (PCE) of 8 %. This research provides valuable insights for the development of efficient D-A compounds for use in organic solar cells.</div></div>\",\"PeriodicalId\":284,\"journal\":{\"name\":\"Computational and Theoretical Chemistry\",\"volume\":\"1241 \",\"pages\":\"Article 114899\"},\"PeriodicalIF\":3.0000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computational and Theoretical Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2210271X24004389\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/29 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q3\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computational and Theoretical Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2210271X24004389","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/29 0:00:00","PubModel":"Epub","JCR":"Q3","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Computational study of new small molecules D-A based on triphenylamines for bulk heterojunction solar cells (BHJ)
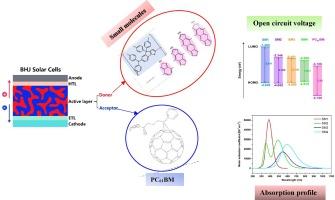
In this computational study, four donor–acceptor (D–A) small molecules were constructed using indacenodithiophene triphenylamine (IDTTPA) as the donor, with modifications made to the acceptor units intended for use in organic bulk heterojunction (BHJ) solar cells. Their electrical and optical properties were determined through density functional theory (DFT) and time-dependent DFT (TD-DFT) techniques. To ascertain the impact of acceptor group modifications on intramolecular electron delocalization and light absorption capabilities, several key variables were examined. Based on the results, it was determined that molecule SM4 demonstrated the best performance among the designed compounds. It exhibited a maximum wavelength of 594 nm, a narrow energy gap of 1.91 eV, a low-lying HOMO energy level of −4.512 eV in its absorption spectra, and a theoretical power conversion efficiency (PCE) of 8 %. This research provides valuable insights for the development of efficient D-A compounds for use in organic solar cells.
期刊介绍:
Computational and Theoretical Chemistry publishes high quality, original reports of significance in computational and theoretical chemistry including those that deal with problems of structure, properties, energetics, weak interactions, reaction mechanisms, catalysis, and reaction rates involving atoms, molecules, clusters, surfaces, and bulk matter.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
