Linyi Zheng, Qian Shen, Guanghong Fang, Ian J Robertson, Qiqiang Long
{"title":"套细胞淋巴瘤中硼替佐米耐药相关蛋白和信号通路的生物信息学研究。","authors":"Linyi Zheng, Qian Shen, Guanghong Fang, Ian J Robertson, Qiqiang Long","doi":"10.21037/tcr-24-1482","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>The bortezomib (BTZ) resistance mechanisms in mantle cell lymphoma (MCL) are complex, involving various genes and signaling pathways. This study used bioinformatical tools to identify and analyze differentially expressed genes (DEGs) associated with BTZ resistance.</p><p><strong>Methods: </strong>Gene chip datasets containing MCL BTZ-resistant and normal control cohorts (GSE20915 and GSE51371) were selected from the Gene Expression Omnibus (GEO) database. GEO2R was used to identify the upregulated DEGs in the microarray datasets, using a significance threshold of P<0.05. Subsequently, these DEGs were subjected to a Gene Ontology (GO) functional analysis, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and a protein-protein interaction (PPI) network assessment. Additionally, 40 MCL patients who underwent second-line BTZ treatment were included in this study. The patients were categorized into resistant and sensitive groups based on treatment response. The enzyme-linked immunosorbent assay (ELISA) technique was employed to evaluate the expression levels of specific DEGs in the serum of the patients in both groups.</p><p><strong>Results: </strong>In the GSE20915 dataset, 144 upregulated genes were identified as DEGs. Similarly, in the GSE51371 dataset, 219 upregulated genes were identified as DEGs. By employing a Venn diagram to compare the upregulated DEGs from both datasets, we identified 11 DEGs linked to BTZ resistance in MCL. The enrichment analysis of the KEGG signaling pathways revealed that the DEGs were predominantly enriched in key biological processes (BP), including the cell cycle, cellular senescence, the p53 signaling pathway, the interleukin 17 (IL-17) signaling pathway, and the nuclear factor kappa-B (NF-κB) signaling pathway. A distinct cluster was revealed by creating a PPI network and performing a module analysis of a set of typical DEGs. This cluster comprised four candidate genes; that is, cyclin-dependent kinase inhibitor 1A (<i>CDKN1A</i>), <i>CDKN1C</i>, midkine (<i>MDK</i>), and TNF alpha induced protein 3 (<i>TNFAIP3</i>). Among these genes, <i>MDK</i> was found to be the key gene. The serum concentration of <i>MDK</i> in the resistant group [1,539 (1,212, 2,023) ng/L] was significantly higher than that in the sensitive group [1,175 (786, 1,502) ng/L] (P<0.05).</p><p><strong>Conclusion: </strong>Identifying the key gene <i>MDK</i> and its associated signaling pathways extends our understanding of the molecular processes that underlie resistance to BTZ in MCL. This discovery establishes a theoretical framework for future investigations of targeted therapy in clinical settings.</p>","PeriodicalId":23216,"journal":{"name":"Translational cancer research","volume":"13 9","pages":"5087-5096"},"PeriodicalIF":2.1000,"publicationDate":"2024-09-30","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11483405/pdf/","citationCount":"0","resultStr":"{\"title\":\"Bioinformatics study of bortezomib resistance-related proteins and signaling pathways in mantle cell lymphoma.\",\"authors\":\"Linyi Zheng, Qian Shen, Guanghong Fang, Ian J Robertson, Qiqiang Long\",\"doi\":\"10.21037/tcr-24-1482\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>The bortezomib (BTZ) resistance mechanisms in mantle cell lymphoma (MCL) are complex, involving various genes and signaling pathways. This study used bioinformatical tools to identify and analyze differentially expressed genes (DEGs) associated with BTZ resistance.</p><p><strong>Methods: </strong>Gene chip datasets containing MCL BTZ-resistant and normal control cohorts (GSE20915 and GSE51371) were selected from the Gene Expression Omnibus (GEO) database. GEO2R was used to identify the upregulated DEGs in the microarray datasets, using a significance threshold of P<0.05. Subsequently, these DEGs were subjected to a Gene Ontology (GO) functional analysis, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and a protein-protein interaction (PPI) network assessment. Additionally, 40 MCL patients who underwent second-line BTZ treatment were included in this study. The patients were categorized into resistant and sensitive groups based on treatment response. The enzyme-linked immunosorbent assay (ELISA) technique was employed to evaluate the expression levels of specific DEGs in the serum of the patients in both groups.</p><p><strong>Results: </strong>In the GSE20915 dataset, 144 upregulated genes were identified as DEGs. Similarly, in the GSE51371 dataset, 219 upregulated genes were identified as DEGs. By employing a Venn diagram to compare the upregulated DEGs from both datasets, we identified 11 DEGs linked to BTZ resistance in MCL. The enrichment analysis of the KEGG signaling pathways revealed that the DEGs were predominantly enriched in key biological processes (BP), including the cell cycle, cellular senescence, the p53 signaling pathway, the interleukin 17 (IL-17) signaling pathway, and the nuclear factor kappa-B (NF-κB) signaling pathway. A distinct cluster was revealed by creating a PPI network and performing a module analysis of a set of typical DEGs. This cluster comprised four candidate genes; that is, cyclin-dependent kinase inhibitor 1A (<i>CDKN1A</i>), <i>CDKN1C</i>, midkine (<i>MDK</i>), and TNF alpha induced protein 3 (<i>TNFAIP3</i>). Among these genes, <i>MDK</i> was found to be the key gene. The serum concentration of <i>MDK</i> in the resistant group [1,539 (1,212, 2,023) ng/L] was significantly higher than that in the sensitive group [1,175 (786, 1,502) ng/L] (P<0.05).</p><p><strong>Conclusion: </strong>Identifying the key gene <i>MDK</i> and its associated signaling pathways extends our understanding of the molecular processes that underlie resistance to BTZ in MCL. This discovery establishes a theoretical framework for future investigations of targeted therapy in clinical settings.</p>\",\"PeriodicalId\":23216,\"journal\":{\"name\":\"Translational cancer research\",\"volume\":\"13 9\",\"pages\":\"5087-5096\"},\"PeriodicalIF\":2.1000,\"publicationDate\":\"2024-09-30\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11483405/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Translational cancer research\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.21037/tcr-24-1482\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/27 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q4\",\"JCRName\":\"ONCOLOGY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Translational cancer research","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.21037/tcr-24-1482","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/27 0:00:00","PubModel":"Epub","JCR":"Q4","JCRName":"ONCOLOGY","Score":null,"Total":0}
Bioinformatics study of bortezomib resistance-related proteins and signaling pathways in mantle cell lymphoma.
Background: The bortezomib (BTZ) resistance mechanisms in mantle cell lymphoma (MCL) are complex, involving various genes and signaling pathways. This study used bioinformatical tools to identify and analyze differentially expressed genes (DEGs) associated with BTZ resistance.
Methods: Gene chip datasets containing MCL BTZ-resistant and normal control cohorts (GSE20915 and GSE51371) were selected from the Gene Expression Omnibus (GEO) database. GEO2R was used to identify the upregulated DEGs in the microarray datasets, using a significance threshold of P<0.05. Subsequently, these DEGs were subjected to a Gene Ontology (GO) functional analysis, a Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis, and a protein-protein interaction (PPI) network assessment. Additionally, 40 MCL patients who underwent second-line BTZ treatment were included in this study. The patients were categorized into resistant and sensitive groups based on treatment response. The enzyme-linked immunosorbent assay (ELISA) technique was employed to evaluate the expression levels of specific DEGs in the serum of the patients in both groups.
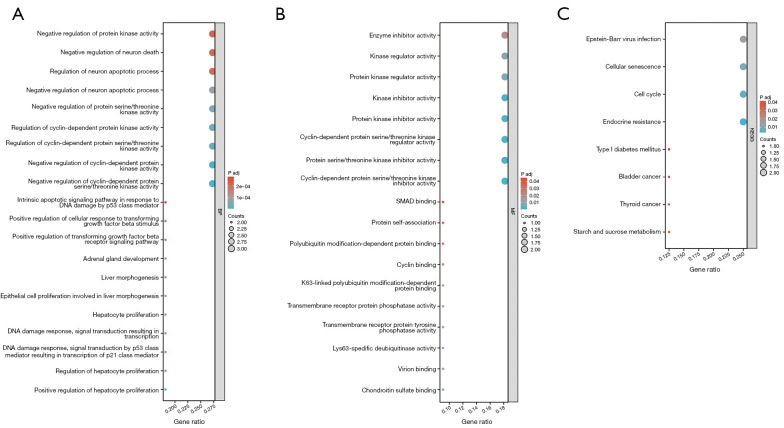
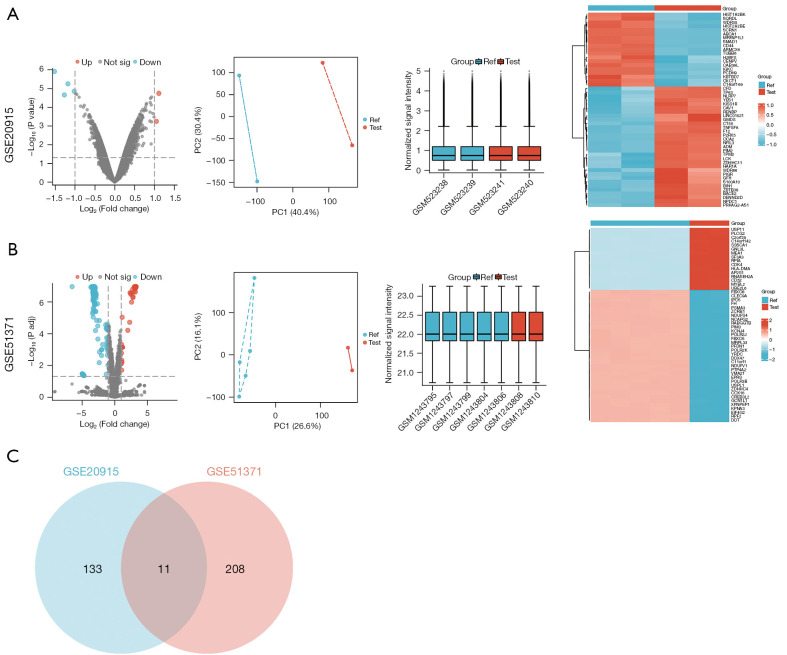
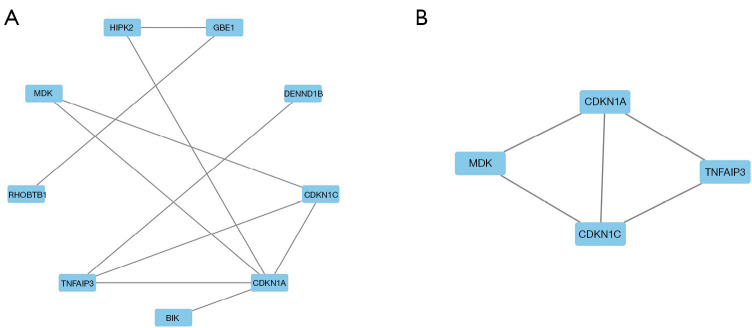
Results: In the GSE20915 dataset, 144 upregulated genes were identified as DEGs. Similarly, in the GSE51371 dataset, 219 upregulated genes were identified as DEGs. By employing a Venn diagram to compare the upregulated DEGs from both datasets, we identified 11 DEGs linked to BTZ resistance in MCL. The enrichment analysis of the KEGG signaling pathways revealed that the DEGs were predominantly enriched in key biological processes (BP), including the cell cycle, cellular senescence, the p53 signaling pathway, the interleukin 17 (IL-17) signaling pathway, and the nuclear factor kappa-B (NF-κB) signaling pathway. A distinct cluster was revealed by creating a PPI network and performing a module analysis of a set of typical DEGs. This cluster comprised four candidate genes; that is, cyclin-dependent kinase inhibitor 1A (CDKN1A), CDKN1C, midkine (MDK), and TNF alpha induced protein 3 (TNFAIP3). Among these genes, MDK was found to be the key gene. The serum concentration of MDK in the resistant group [1,539 (1,212, 2,023) ng/L] was significantly higher than that in the sensitive group [1,175 (786, 1,502) ng/L] (P<0.05).
Conclusion: Identifying the key gene MDK and its associated signaling pathways extends our understanding of the molecular processes that underlie resistance to BTZ in MCL. This discovery establishes a theoretical framework for future investigations of targeted therapy in clinical settings.
期刊介绍:
Translational Cancer Research (Transl Cancer Res TCR; Print ISSN: 2218-676X; Online ISSN 2219-6803; http://tcr.amegroups.com/) is an Open Access, peer-reviewed journal, indexed in Science Citation Index Expanded (SCIE). TCR publishes laboratory studies of novel therapeutic interventions as well as clinical trials which evaluate new treatment paradigms for cancer; results of novel research investigations which bridge the laboratory and clinical settings including risk assessment, cellular and molecular characterization, prevention, detection, diagnosis and treatment of human cancers with the overall goal of improving the clinical care of cancer patients. The focus of TCR is original, peer-reviewed, science-based research that successfully advances clinical medicine toward the goal of improving patients'' quality of life. The editors and an international advisory group of scientists and clinician-scientists as well as other experts will hold TCR articles to the high-quality standards. We accept Original Articles as well as Review Articles, Editorials and Brief Articles.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
