Haochen Ning, Ian Boyes, Ibrahim Numanagić, Michael Rott, Li Xing, Xuekui Zhang
{"title":"利用高通量基因组测序数据诊断病毒感染。","authors":"Haochen Ning, Ian Boyes, Ibrahim Numanagić, Michael Rott, Li Xing, Xuekui Zhang","doi":"10.1093/bib/bbae501","DOIUrl":null,"url":null,"abstract":"<p><p>Plant viral infections cause significant economic losses, totalling $350 billion USD in 2021. With no treatment for virus-infected plants, accurate and efficient diagnosis is crucial to preventing and controlling these diseases. High-throughput sequencing (HTS) enables cost-efficient identification of known and unknown viruses. However, existing diagnostic pipelines face challenges. First, many methods depend on subjectively chosen parameter values, undermining their robustness across various data sources. Second, artifacts (e.g. false peaks) in the mapped sequence data can lead to incorrect diagnostic results. While some methods require manual or subjective verification to address these artifacts, others overlook them entirely, affecting the overall method performance and leading to imprecise or labour-intensive outcomes. To address these challenges, we introduce IIMI, a new automated analysis pipeline using machine learning to diagnose infections from 1583 plant viruses with HTS data. It adopts a data-driven approach for parameter selection, reducing subjectivity, and automatically filters out regions affected by artifacts, thus improving accuracy. Testing with in-house and published data shows IIMI's superiority over existing methods. Besides a prediction model, IIMI also provides resources on plant virus genomes, including annotations of regions prone to artifacts. The method is available as an R package (iimi) on CRAN and will integrate with the web application www.virtool.ca, enhancing accessibility and user convenience.</p>","PeriodicalId":9209,"journal":{"name":"Briefings in bioinformatics","volume":"25 6","pages":""},"PeriodicalIF":7.7000,"publicationDate":"2024-09-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11483527/pdf/","citationCount":"0","resultStr":"{\"title\":\"Diagnostics of viral infections using high-throughput genome sequencing data.\",\"authors\":\"Haochen Ning, Ian Boyes, Ibrahim Numanagić, Michael Rott, Li Xing, Xuekui Zhang\",\"doi\":\"10.1093/bib/bbae501\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>Plant viral infections cause significant economic losses, totalling $350 billion USD in 2021. With no treatment for virus-infected plants, accurate and efficient diagnosis is crucial to preventing and controlling these diseases. High-throughput sequencing (HTS) enables cost-efficient identification of known and unknown viruses. However, existing diagnostic pipelines face challenges. First, many methods depend on subjectively chosen parameter values, undermining their robustness across various data sources. Second, artifacts (e.g. false peaks) in the mapped sequence data can lead to incorrect diagnostic results. While some methods require manual or subjective verification to address these artifacts, others overlook them entirely, affecting the overall method performance and leading to imprecise or labour-intensive outcomes. To address these challenges, we introduce IIMI, a new automated analysis pipeline using machine learning to diagnose infections from 1583 plant viruses with HTS data. It adopts a data-driven approach for parameter selection, reducing subjectivity, and automatically filters out regions affected by artifacts, thus improving accuracy. Testing with in-house and published data shows IIMI's superiority over existing methods. Besides a prediction model, IIMI also provides resources on plant virus genomes, including annotations of regions prone to artifacts. The method is available as an R package (iimi) on CRAN and will integrate with the web application www.virtool.ca, enhancing accessibility and user convenience.</p>\",\"PeriodicalId\":9209,\"journal\":{\"name\":\"Briefings in bioinformatics\",\"volume\":\"25 6\",\"pages\":\"\"},\"PeriodicalIF\":7.7000,\"publicationDate\":\"2024-09-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11483527/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Briefings in bioinformatics\",\"FirstCategoryId\":\"99\",\"ListUrlMain\":\"https://doi.org/10.1093/bib/bbae501\",\"RegionNum\":2,\"RegionCategory\":\"生物学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"BIOCHEMICAL RESEARCH METHODS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Briefings in bioinformatics","FirstCategoryId":"99","ListUrlMain":"https://doi.org/10.1093/bib/bbae501","RegionNum":2,"RegionCategory":"生物学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"BIOCHEMICAL RESEARCH METHODS","Score":null,"Total":0}
Diagnostics of viral infections using high-throughput genome sequencing data.
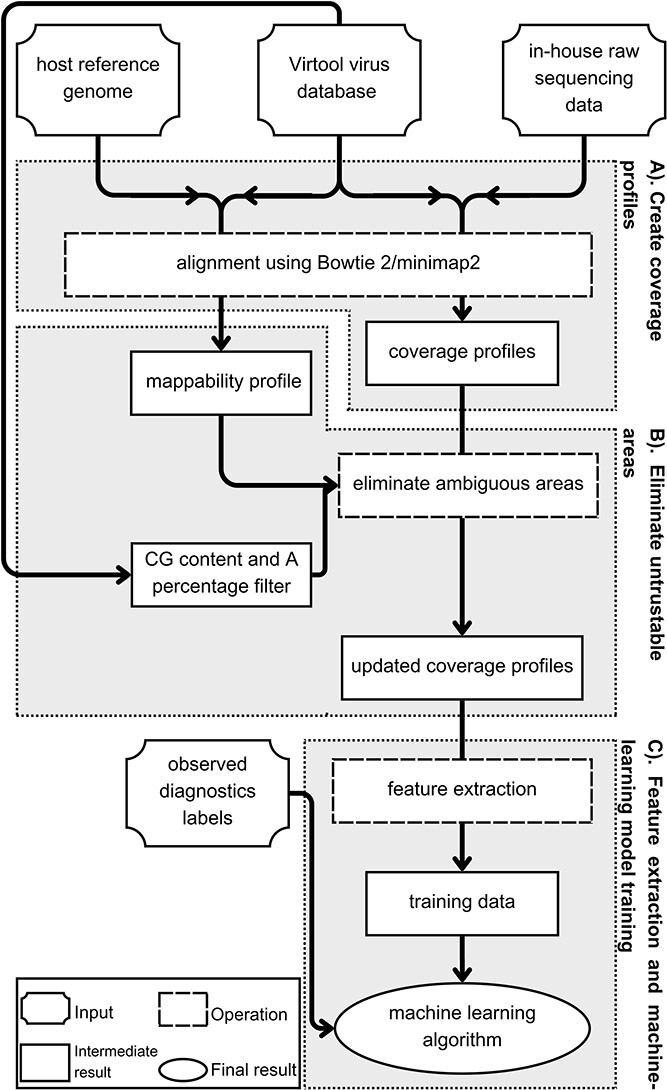
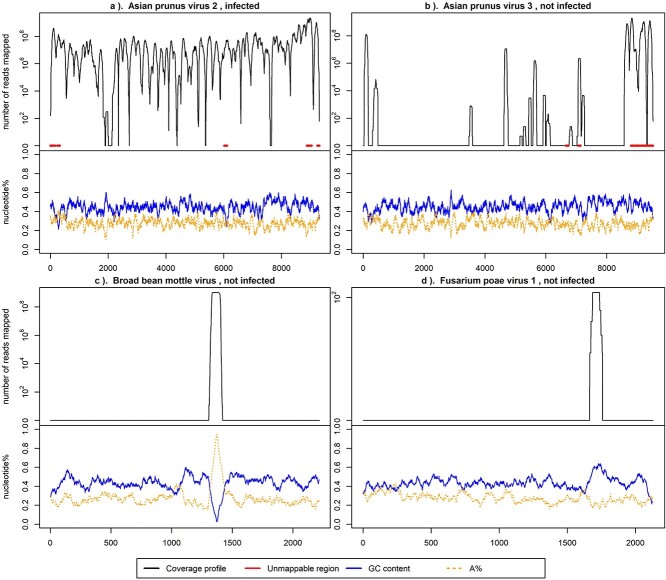
Plant viral infections cause significant economic losses, totalling $350 billion USD in 2021. With no treatment for virus-infected plants, accurate and efficient diagnosis is crucial to preventing and controlling these diseases. High-throughput sequencing (HTS) enables cost-efficient identification of known and unknown viruses. However, existing diagnostic pipelines face challenges. First, many methods depend on subjectively chosen parameter values, undermining their robustness across various data sources. Second, artifacts (e.g. false peaks) in the mapped sequence data can lead to incorrect diagnostic results. While some methods require manual or subjective verification to address these artifacts, others overlook them entirely, affecting the overall method performance and leading to imprecise or labour-intensive outcomes. To address these challenges, we introduce IIMI, a new automated analysis pipeline using machine learning to diagnose infections from 1583 plant viruses with HTS data. It adopts a data-driven approach for parameter selection, reducing subjectivity, and automatically filters out regions affected by artifacts, thus improving accuracy. Testing with in-house and published data shows IIMI's superiority over existing methods. Besides a prediction model, IIMI also provides resources on plant virus genomes, including annotations of regions prone to artifacts. The method is available as an R package (iimi) on CRAN and will integrate with the web application www.virtool.ca, enhancing accessibility and user convenience.
期刊介绍:
Briefings in Bioinformatics is an international journal serving as a platform for researchers and educators in the life sciences. It also appeals to mathematicians, statisticians, and computer scientists applying their expertise to biological challenges. The journal focuses on reviews tailored for users of databases and analytical tools in contemporary genetics, molecular and systems biology. It stands out by offering practical assistance and guidance to non-specialists in computerized methodologies. Covering a wide range from introductory concepts to specific protocols and analyses, the papers address bacterial, plant, fungal, animal, and human data.
The journal's detailed subject areas include genetic studies of phenotypes and genotypes, mapping, DNA sequencing, expression profiling, gene expression studies, microarrays, alignment methods, protein profiles and HMMs, lipids, metabolic and signaling pathways, structure determination and function prediction, phylogenetic studies, and education and training.





 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
