{"title":"利用 DOCK 构建蛋白质中有机小分子的片段和扭转偏置算法","authors":"John D. Bickel, Brock T. Boysan, Robert C. Rizzo","doi":"10.1002/jcc.27508","DOIUrl":null,"url":null,"abstract":"<p>The computational construction of small organic molecules (de novo design), directly in a protein binding site, is an effective means for generating novel ligands tailored to fit the pocket environment. In this work, we present two new methods, which aim to improve de novo design outcomes using (1) biasing algorithms to prioritize selection and/or acceptance of fragments and torsions during growth, and (2) parallel-based clustering and pruning algorithms to remove duplicate molecules as candidate fragment are added. Large-scale testing encompassing thousands of simulations were employed to interrogate the methods in terms of multiple metrics which include numbers of duplicate molecules generated, pairwise-similarity, focused library reconstruction rates, fragment and torsion frequencies, fragment and torsion rank scores, interaction energy and drug-likeness scores, and 3D pose comparisons. The biasing algorithms, particularly those that include fragment and torsion components simultaneously, led to molecules that more closely mimicked the distributions of fragments and torsions found in drug-like libraries. The new parallel-based clustering and pruning algorithms, compared with the existing serial approach, also led to larger ensembles comprised of topologically unique molecules with much greater efficiency by removing redundant growth paths.</p>","PeriodicalId":188,"journal":{"name":"Journal of Computational Chemistry","volume":"46 1","pages":""},"PeriodicalIF":4.8000,"publicationDate":"2024-10-22","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Fragment and torsion biasing algorithms for construction of small organic molecules in proteins using DOCK\",\"authors\":\"John D. Bickel, Brock T. Boysan, Robert C. Rizzo\",\"doi\":\"10.1002/jcc.27508\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p>The computational construction of small organic molecules (de novo design), directly in a protein binding site, is an effective means for generating novel ligands tailored to fit the pocket environment. In this work, we present two new methods, which aim to improve de novo design outcomes using (1) biasing algorithms to prioritize selection and/or acceptance of fragments and torsions during growth, and (2) parallel-based clustering and pruning algorithms to remove duplicate molecules as candidate fragment are added. Large-scale testing encompassing thousands of simulations were employed to interrogate the methods in terms of multiple metrics which include numbers of duplicate molecules generated, pairwise-similarity, focused library reconstruction rates, fragment and torsion frequencies, fragment and torsion rank scores, interaction energy and drug-likeness scores, and 3D pose comparisons. The biasing algorithms, particularly those that include fragment and torsion components simultaneously, led to molecules that more closely mimicked the distributions of fragments and torsions found in drug-like libraries. The new parallel-based clustering and pruning algorithms, compared with the existing serial approach, also led to larger ensembles comprised of topologically unique molecules with much greater efficiency by removing redundant growth paths.</p>\",\"PeriodicalId\":188,\"journal\":{\"name\":\"Journal of Computational Chemistry\",\"volume\":\"46 1\",\"pages\":\"\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-10-22\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Computational Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27508\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, MULTIDISCIPLINARY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Computational Chemistry","FirstCategoryId":"92","ListUrlMain":"https://onlinelibrary.wiley.com/doi/10.1002/jcc.27508","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, MULTIDISCIPLINARY","Score":null,"Total":0}
Fragment and torsion biasing algorithms for construction of small organic molecules in proteins using DOCK
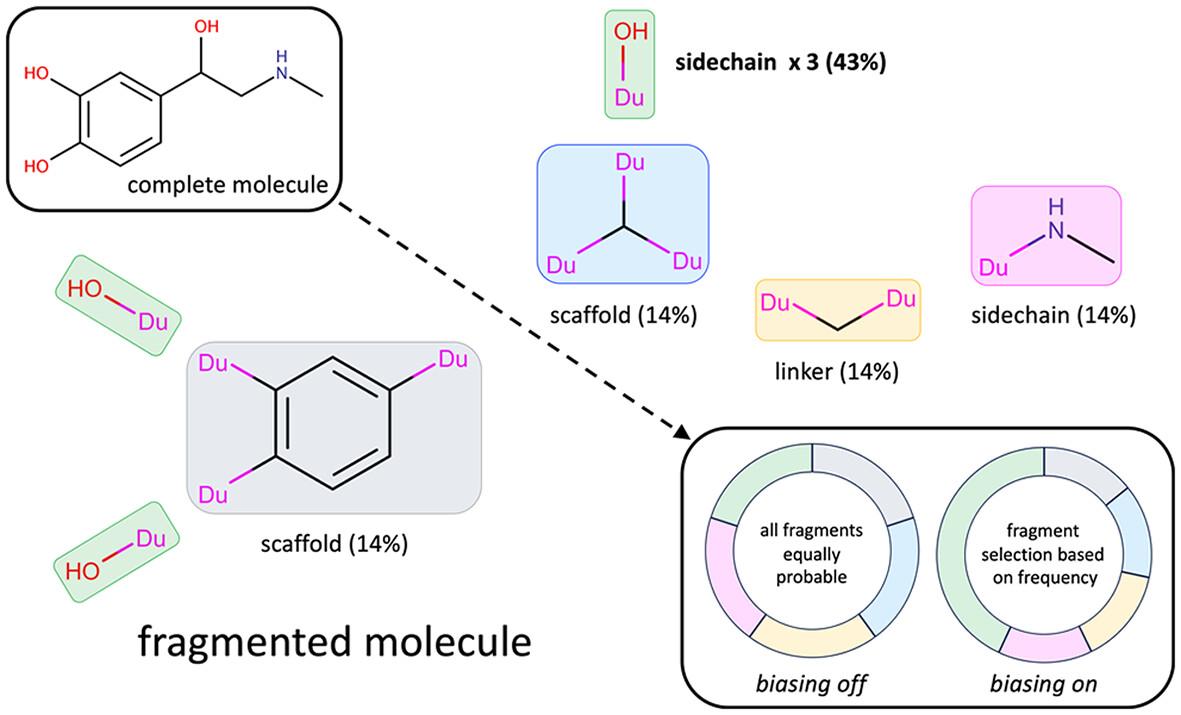
The computational construction of small organic molecules (de novo design), directly in a protein binding site, is an effective means for generating novel ligands tailored to fit the pocket environment. In this work, we present two new methods, which aim to improve de novo design outcomes using (1) biasing algorithms to prioritize selection and/or acceptance of fragments and torsions during growth, and (2) parallel-based clustering and pruning algorithms to remove duplicate molecules as candidate fragment are added. Large-scale testing encompassing thousands of simulations were employed to interrogate the methods in terms of multiple metrics which include numbers of duplicate molecules generated, pairwise-similarity, focused library reconstruction rates, fragment and torsion frequencies, fragment and torsion rank scores, interaction energy and drug-likeness scores, and 3D pose comparisons. The biasing algorithms, particularly those that include fragment and torsion components simultaneously, led to molecules that more closely mimicked the distributions of fragments and torsions found in drug-like libraries. The new parallel-based clustering and pruning algorithms, compared with the existing serial approach, also led to larger ensembles comprised of topologically unique molecules with much greater efficiency by removing redundant growth paths.
期刊介绍:
This distinguished journal publishes articles concerned with all aspects of computational chemistry: analytical, biological, inorganic, organic, physical, and materials. The Journal of Computational Chemistry presents original research, contemporary developments in theory and methodology, and state-of-the-art applications. Computational areas that are featured in the journal include ab initio and semiempirical quantum mechanics, density functional theory, molecular mechanics, molecular dynamics, statistical mechanics, cheminformatics, biomolecular structure prediction, molecular design, and bioinformatics.



 分享
分享

 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
