{"title":"加速扩散的 Ab Initio 纳米反应器分子动力学及硫化氢氧化应用","authors":"Jan A. Meissner, Jan Meisner","doi":"10.1021/acs.jctc.4c00826","DOIUrl":null,"url":null,"abstract":"The computational description of chemical reactivity can become extremely complex when multiple different reaction products and intermediates come into play, forming a chemical reaction network. Therefore, computational methods for the automated construction of chemical reaction networks have been developed in the last decades. One of these methods, ab initio nanoreactor molecular dynamics (NMD), is based on external forces enhancing reactivity by e.g., periodically compressing the system and allowing it to relax. However, during the relaxation process, a significant simulation time is required to allow energy to dissipate and molecules to diffuse, making this part of the NMD simulation computationally intensive. This work aims to improve NMD by accelerating the diffusion process in the relaxation phase. We systematically investigate the speedup of reaction discovery gained by diffusion acceleration, leading to a factor of up to 28 in discovery frequency. Diffusion-accelerated nanoreactor molecular dynamics (DA-NMD) is then used to construct a reaction network of hydrogen sulfide oxidation under atmospheric conditions, where reactions are automatically detected by a change in the bond order and bond distance. A reaction network of 108 molecular species and 399 elementary reactions was constructed starting from hydrogen sulfide, hydroxy radicals, and molecular oxygen covering a broad variety of sulfur–oxygen chemistry and oxidation states of the sulfur atom ranging from −II to +VI.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":"33 1","pages":""},"PeriodicalIF":5.5000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Acceleration of Diffusion in Ab Initio Nanoreactor Molecular Dynamics and Application to Hydrogen Sulfide Oxidation\",\"authors\":\"Jan A. Meissner, Jan Meisner\",\"doi\":\"10.1021/acs.jctc.4c00826\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"The computational description of chemical reactivity can become extremely complex when multiple different reaction products and intermediates come into play, forming a chemical reaction network. Therefore, computational methods for the automated construction of chemical reaction networks have been developed in the last decades. One of these methods, ab initio nanoreactor molecular dynamics (NMD), is based on external forces enhancing reactivity by e.g., periodically compressing the system and allowing it to relax. However, during the relaxation process, a significant simulation time is required to allow energy to dissipate and molecules to diffuse, making this part of the NMD simulation computationally intensive. This work aims to improve NMD by accelerating the diffusion process in the relaxation phase. We systematically investigate the speedup of reaction discovery gained by diffusion acceleration, leading to a factor of up to 28 in discovery frequency. Diffusion-accelerated nanoreactor molecular dynamics (DA-NMD) is then used to construct a reaction network of hydrogen sulfide oxidation under atmospheric conditions, where reactions are automatically detected by a change in the bond order and bond distance. A reaction network of 108 molecular species and 399 elementary reactions was constructed starting from hydrogen sulfide, hydroxy radicals, and molecular oxygen covering a broad variety of sulfur–oxygen chemistry and oxidation states of the sulfur atom ranging from −II to +VI.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":\"33 1\",\"pages\":\"\"},\"PeriodicalIF\":5.5000,\"publicationDate\":\"2024-10-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c00826\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c00826","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Acceleration of Diffusion in Ab Initio Nanoreactor Molecular Dynamics and Application to Hydrogen Sulfide Oxidation
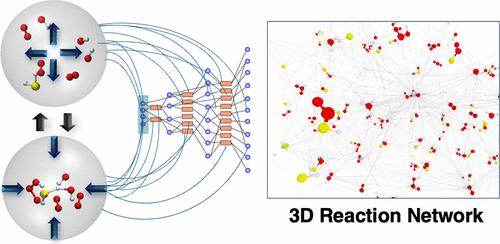
The computational description of chemical reactivity can become extremely complex when multiple different reaction products and intermediates come into play, forming a chemical reaction network. Therefore, computational methods for the automated construction of chemical reaction networks have been developed in the last decades. One of these methods, ab initio nanoreactor molecular dynamics (NMD), is based on external forces enhancing reactivity by e.g., periodically compressing the system and allowing it to relax. However, during the relaxation process, a significant simulation time is required to allow energy to dissipate and molecules to diffuse, making this part of the NMD simulation computationally intensive. This work aims to improve NMD by accelerating the diffusion process in the relaxation phase. We systematically investigate the speedup of reaction discovery gained by diffusion acceleration, leading to a factor of up to 28 in discovery frequency. Diffusion-accelerated nanoreactor molecular dynamics (DA-NMD) is then used to construct a reaction network of hydrogen sulfide oxidation under atmospheric conditions, where reactions are automatically detected by a change in the bond order and bond distance. A reaction network of 108 molecular species and 399 elementary reactions was constructed starting from hydrogen sulfide, hydroxy radicals, and molecular oxygen covering a broad variety of sulfur–oxygen chemistry and oxidation states of the sulfur atom ranging from −II to +VI.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
