{"title":"利用基于生成变压器架构的神经网络精确计算原子间作用力","authors":"Juntao Lai, Bowen Kan, Yangjun Wu, Qiang Fu, Honghui Shang, Zhenyu Li, Jinlong Yang","doi":"10.1021/acs.jctc.4c01205","DOIUrl":null,"url":null,"abstract":"Using neural networks to express electronic wave functions represents a new paradigm for solving the Schrödinger equation in quantum chemistry. For practical quantum chemistry simulations, one needs to know not only energies of molecules, but also accurate forces acting on constituent atoms. In this work, we achieve the accurate calculation of interatomic forces on QiankunNet, a platform that combines transformer-based deep neural networks with efficient batched autoregressive sampling. Our approach permits the application of the Hellmann–Feynman theorem to force calculations without introducing corrective Pulay terms. The results show that the calculated interatomic forces are in close agreement with those derived from the full configuration interaction method, irrespective of whether the system is a simple molecule or a strongly correlated electron system like a linear hydrogen chain. Furthermore, the calculated interatomic forces are employed for atomic relaxation in the torsional rotation process of ethylene, and the energy barrier obtained from the scanned potential energy surface is in excellent agreement with the experiment. Our work contributes to the application of artificial intelligence to broader quantum chemistry simulations, such as modeling challenging chemical transformations where electron correlations are difficult to describe.","PeriodicalId":45,"journal":{"name":"Journal of Chemical Theory and Computation","volume":null,"pages":null},"PeriodicalIF":5.7000,"publicationDate":"2024-10-23","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Accurate Calculation of Interatomic Forces with Neural Networks Based on a Generative Transformer Architecture\",\"authors\":\"Juntao Lai, Bowen Kan, Yangjun Wu, Qiang Fu, Honghui Shang, Zhenyu Li, Jinlong Yang\",\"doi\":\"10.1021/acs.jctc.4c01205\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"Using neural networks to express electronic wave functions represents a new paradigm for solving the Schrödinger equation in quantum chemistry. For practical quantum chemistry simulations, one needs to know not only energies of molecules, but also accurate forces acting on constituent atoms. In this work, we achieve the accurate calculation of interatomic forces on QiankunNet, a platform that combines transformer-based deep neural networks with efficient batched autoregressive sampling. Our approach permits the application of the Hellmann–Feynman theorem to force calculations without introducing corrective Pulay terms. The results show that the calculated interatomic forces are in close agreement with those derived from the full configuration interaction method, irrespective of whether the system is a simple molecule or a strongly correlated electron system like a linear hydrogen chain. Furthermore, the calculated interatomic forces are employed for atomic relaxation in the torsional rotation process of ethylene, and the energy barrier obtained from the scanned potential energy surface is in excellent agreement with the experiment. Our work contributes to the application of artificial intelligence to broader quantum chemistry simulations, such as modeling challenging chemical transformations where electron correlations are difficult to describe.\",\"PeriodicalId\":45,\"journal\":{\"name\":\"Journal of Chemical Theory and Computation\",\"volume\":null,\"pages\":null},\"PeriodicalIF\":5.7000,\"publicationDate\":\"2024-10-23\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Chemical Theory and Computation\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jctc.4c01205\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Chemical Theory and Computation","FirstCategoryId":"92","ListUrlMain":"https://doi.org/10.1021/acs.jctc.4c01205","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
Accurate Calculation of Interatomic Forces with Neural Networks Based on a Generative Transformer Architecture
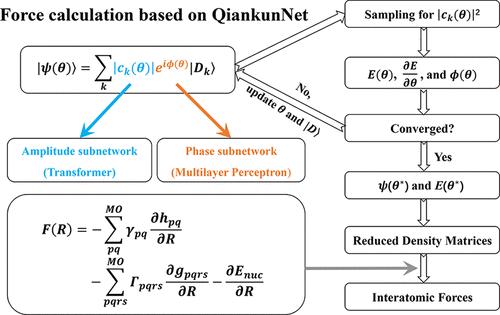
Using neural networks to express electronic wave functions represents a new paradigm for solving the Schrödinger equation in quantum chemistry. For practical quantum chemistry simulations, one needs to know not only energies of molecules, but also accurate forces acting on constituent atoms. In this work, we achieve the accurate calculation of interatomic forces on QiankunNet, a platform that combines transformer-based deep neural networks with efficient batched autoregressive sampling. Our approach permits the application of the Hellmann–Feynman theorem to force calculations without introducing corrective Pulay terms. The results show that the calculated interatomic forces are in close agreement with those derived from the full configuration interaction method, irrespective of whether the system is a simple molecule or a strongly correlated electron system like a linear hydrogen chain. Furthermore, the calculated interatomic forces are employed for atomic relaxation in the torsional rotation process of ethylene, and the energy barrier obtained from the scanned potential energy surface is in excellent agreement with the experiment. Our work contributes to the application of artificial intelligence to broader quantum chemistry simulations, such as modeling challenging chemical transformations where electron correlations are difficult to describe.
期刊介绍:
The Journal of Chemical Theory and Computation invites new and original contributions with the understanding that, if accepted, they will not be published elsewhere. Papers reporting new theories, methodology, and/or important applications in quantum electronic structure, molecular dynamics, and statistical mechanics are appropriate for submission to this Journal. Specific topics include advances in or applications of ab initio quantum mechanics, density functional theory, design and properties of new materials, surface science, Monte Carlo simulations, solvation models, QM/MM calculations, biomolecular structure prediction, and molecular dynamics in the broadest sense including gas-phase dynamics, ab initio dynamics, biomolecular dynamics, and protein folding. The Journal does not consider papers that are straightforward applications of known methods including DFT and molecular dynamics. The Journal favors submissions that include advances in theory or methodology with applications to compelling problems.



 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
