Xin He , Yujie Zhang , Haomiao Li , Min Zhou , Wei Wang , Ruxing Wang , Kai Jiang , Kangli Wang
{"title":"提高经典分子动力学模拟在电池电解质设计中的可靠性","authors":"Xin He , Yujie Zhang , Haomiao Li , Min Zhou , Wei Wang , Ruxing Wang , Kai Jiang , Kangli Wang","doi":"10.1016/j.jechem.2024.09.038","DOIUrl":null,"url":null,"abstract":"<div><div>Explorations into new electrolytes have highlighted the critical impact of solvation structure on battery performance. Classical molecular dynamics (CMD) using semi-empirical force fields has become an essential tool for simulating solvation structures. However, mainstream force fields often lack accuracy in describing strong ion-solvent interactions, causing disparities between CMD simulations and experimental observations. Although some empirical methods have been employed in some of the studies to address this issue, their effectiveness has been limited. Our CMD research, supported by quantum chemical calculations and experimental data, reveals that the solvation structure is influenced not only by the charge model but also by the polarization description. Previous empirical approaches that focused solely on adjusting ion-solvent interaction strengths overlooked the importance of polarization effects. Building on this insight, we propose integrating the Drude polarization model into mainstream force fields and verify its feasibility in carbonate, ether, and nitrile electrolytes. Our experimental results demonstrate that this approach significantly enhances the accuracy of CMD-simulated solvation structures. This work is expected to provide a more reliable CMD method for electrolyte design, shielding researchers from the pitfalls of erroneous simulation outcomes.</div></div>","PeriodicalId":15728,"journal":{"name":"Journal of Energy Chemistry","volume":"101 ","pages":"Pages 34-41"},"PeriodicalIF":15.6000,"publicationDate":"2025-02-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Improving the reliability of classical molecular dynamics simulations in battery electrolyte design\",\"authors\":\"Xin He , Yujie Zhang , Haomiao Li , Min Zhou , Wei Wang , Ruxing Wang , Kai Jiang , Kangli Wang\",\"doi\":\"10.1016/j.jechem.2024.09.038\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><div>Explorations into new electrolytes have highlighted the critical impact of solvation structure on battery performance. Classical molecular dynamics (CMD) using semi-empirical force fields has become an essential tool for simulating solvation structures. However, mainstream force fields often lack accuracy in describing strong ion-solvent interactions, causing disparities between CMD simulations and experimental observations. Although some empirical methods have been employed in some of the studies to address this issue, their effectiveness has been limited. Our CMD research, supported by quantum chemical calculations and experimental data, reveals that the solvation structure is influenced not only by the charge model but also by the polarization description. Previous empirical approaches that focused solely on adjusting ion-solvent interaction strengths overlooked the importance of polarization effects. Building on this insight, we propose integrating the Drude polarization model into mainstream force fields and verify its feasibility in carbonate, ether, and nitrile electrolytes. Our experimental results demonstrate that this approach significantly enhances the accuracy of CMD-simulated solvation structures. This work is expected to provide a more reliable CMD method for electrolyte design, shielding researchers from the pitfalls of erroneous simulation outcomes.</div></div>\",\"PeriodicalId\":15728,\"journal\":{\"name\":\"Journal of Energy Chemistry\",\"volume\":\"101 \",\"pages\":\"Pages 34-41\"},\"PeriodicalIF\":15.6000,\"publicationDate\":\"2025-02-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Journal of Energy Chemistry\",\"FirstCategoryId\":\"92\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S2095495624006624\",\"RegionNum\":1,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/9/30 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"Energy\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Journal of Energy Chemistry","FirstCategoryId":"92","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S2095495624006624","RegionNum":1,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/9/30 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"Energy","Score":null,"Total":0}
Improving the reliability of classical molecular dynamics simulations in battery electrolyte design
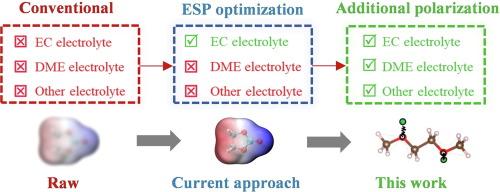
Explorations into new electrolytes have highlighted the critical impact of solvation structure on battery performance. Classical molecular dynamics (CMD) using semi-empirical force fields has become an essential tool for simulating solvation structures. However, mainstream force fields often lack accuracy in describing strong ion-solvent interactions, causing disparities between CMD simulations and experimental observations. Although some empirical methods have been employed in some of the studies to address this issue, their effectiveness has been limited. Our CMD research, supported by quantum chemical calculations and experimental data, reveals that the solvation structure is influenced not only by the charge model but also by the polarization description. Previous empirical approaches that focused solely on adjusting ion-solvent interaction strengths overlooked the importance of polarization effects. Building on this insight, we propose integrating the Drude polarization model into mainstream force fields and verify its feasibility in carbonate, ether, and nitrile electrolytes. Our experimental results demonstrate that this approach significantly enhances the accuracy of CMD-simulated solvation structures. This work is expected to provide a more reliable CMD method for electrolyte design, shielding researchers from the pitfalls of erroneous simulation outcomes.
期刊介绍:
The Journal of Energy Chemistry, the official publication of Science Press and the Dalian Institute of Chemical Physics, Chinese Academy of Sciences, serves as a platform for reporting creative research and innovative applications in energy chemistry. It mainly reports on creative researches and innovative applications of chemical conversions of fossil energy, carbon dioxide, electrochemical energy and hydrogen energy, as well as the conversions of biomass and solar energy related with chemical issues to promote academic exchanges in the field of energy chemistry and to accelerate the exploration, research and development of energy science and technologies.
This journal focuses on original research papers covering various topics within energy chemistry worldwide, including:
Optimized utilization of fossil energy
Hydrogen energy
Conversion and storage of electrochemical energy
Capture, storage, and chemical conversion of carbon dioxide
Materials and nanotechnologies for energy conversion and storage
Chemistry in biomass conversion
Chemistry in the utilization of solar energy




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
