Yuwei Zhang, Nadire Nayir, Yun Kyung Shin, Qian Mao, Ga-Un Jeong, Chen Chen, Joan M. Redwing, Adri C. T. van Duin
{"title":"α-Al2O3 基质与 H2O/H2 气相分子间表面化学反应的 ReaxFF 研究","authors":"Yuwei Zhang, Nadire Nayir, Yun Kyung Shin, Qian Mao, Ga-Un Jeong, Chen Chen, Joan M. Redwing, Adri C. T. van Duin","doi":"10.1021/acs.jpcc.4c04669","DOIUrl":null,"url":null,"abstract":"We developed an Al/O/H ReaxFF force field to explore chemical reactions on α-Al<sub>2</sub>O<sub>3</sub> surfaces in H<sub>2</sub>O/H<sub>2</sub> gas-phase environments. This force field generates surface energy profiles of A-, C-, R-, and M-planes with various terminations (Al- or O-) and predicts the thermodynamic and kinetic behaviors of hydrolysis on Al-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001), consistent with quantum chemical studies. Molecular dynamics (MD) simulations of H<sub>2</sub>O/α-Al<sub>2</sub>O<sub>3</sub> (0001) reveal that water autocatalysis plays a significant role in accelerating H<sub>2</sub>O dissociations on Al-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001). Compared with the 50% Al-terminated surface, the 100% Al-terminated surface becomes more easily hydroxylated at temperatures as low as 350 K, relying more on an O<i><sub>x</sub></i>H<i><sub>y</sub></i> clustering mechanism than complete H<sub>2</sub>O dissociations, and desorbs significantly more H<sub>2</sub>O molecules once heated up to 500 K or higher. But heating cannot eliminate surface hydroxyls for either case, and achieving a Gibbsite-like surface by H<sub>2</sub>O exposure is unlikely. H<sub>2</sub>O dissociations on α-Al<sub>2</sub>O<sub>3</sub> (0001) terminated with randomly distributed surface Al species deviate from 1–2 and 1–4 pathways due to irregular vacancy defects, and a random surface appears to be more reactive to H<sub>2</sub>O than the ordered one with the same surface Al coverage. Simulations of H<sub>2</sub>/α-Al<sub>2</sub>O<sub>3</sub> suggest that the combination of a dense surface O coverage and a low thermodynamic surface stability leads to elevated H<sub>2</sub> dissociation kinetics. To accelerate the surface O removals of 100% O-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001) in H<sub>2</sub> gas exposure, we reduced the H–H σ bond energy parameter, equivalent to lowering the H<sub>2</sub> dissociation barrier by ∼ 19.4 kcal/mol during the simulation. After ∼ 1.5 ns, the surface termination became comparable to the 100% Al-terminated one but retained a small quantity of hydroxyls. This force field reveals how the α-Al<sub>2</sub>O<sub>3</sub> crystallographic plane and the surface termination influence the dissociation behaviors of H<sub>2</sub>O/H<sub>2</sub> gas molecules and lays the foundation for future force field developments targeted at thin film epitaxy on sapphire.","PeriodicalId":61,"journal":{"name":"The Journal of Physical Chemistry C","volume":"11 1","pages":""},"PeriodicalIF":3.4000,"publicationDate":"2024-10-29","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"ReaxFF Study of Surface Chemical Reactions between α-Al2O3 Substrates and H2O/H2 Gas-Phase Molecules\",\"authors\":\"Yuwei Zhang, Nadire Nayir, Yun Kyung Shin, Qian Mao, Ga-Un Jeong, Chen Chen, Joan M. Redwing, Adri C. T. van Duin\",\"doi\":\"10.1021/acs.jpcc.4c04669\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"We developed an Al/O/H ReaxFF force field to explore chemical reactions on α-Al<sub>2</sub>O<sub>3</sub> surfaces in H<sub>2</sub>O/H<sub>2</sub> gas-phase environments. This force field generates surface energy profiles of A-, C-, R-, and M-planes with various terminations (Al- or O-) and predicts the thermodynamic and kinetic behaviors of hydrolysis on Al-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001), consistent with quantum chemical studies. Molecular dynamics (MD) simulations of H<sub>2</sub>O/α-Al<sub>2</sub>O<sub>3</sub> (0001) reveal that water autocatalysis plays a significant role in accelerating H<sub>2</sub>O dissociations on Al-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001). Compared with the 50% Al-terminated surface, the 100% Al-terminated surface becomes more easily hydroxylated at temperatures as low as 350 K, relying more on an O<i><sub>x</sub></i>H<i><sub>y</sub></i> clustering mechanism than complete H<sub>2</sub>O dissociations, and desorbs significantly more H<sub>2</sub>O molecules once heated up to 500 K or higher. But heating cannot eliminate surface hydroxyls for either case, and achieving a Gibbsite-like surface by H<sub>2</sub>O exposure is unlikely. H<sub>2</sub>O dissociations on α-Al<sub>2</sub>O<sub>3</sub> (0001) terminated with randomly distributed surface Al species deviate from 1–2 and 1–4 pathways due to irregular vacancy defects, and a random surface appears to be more reactive to H<sub>2</sub>O than the ordered one with the same surface Al coverage. Simulations of H<sub>2</sub>/α-Al<sub>2</sub>O<sub>3</sub> suggest that the combination of a dense surface O coverage and a low thermodynamic surface stability leads to elevated H<sub>2</sub> dissociation kinetics. To accelerate the surface O removals of 100% O-terminated α-Al<sub>2</sub>O<sub>3</sub> (0001) in H<sub>2</sub> gas exposure, we reduced the H–H σ bond energy parameter, equivalent to lowering the H<sub>2</sub> dissociation barrier by ∼ 19.4 kcal/mol during the simulation. After ∼ 1.5 ns, the surface termination became comparable to the 100% Al-terminated one but retained a small quantity of hydroxyls. This force field reveals how the α-Al<sub>2</sub>O<sub>3</sub> crystallographic plane and the surface termination influence the dissociation behaviors of H<sub>2</sub>O/H<sub>2</sub> gas molecules and lays the foundation for future force field developments targeted at thin film epitaxy on sapphire.\",\"PeriodicalId\":61,\"journal\":{\"name\":\"The Journal of Physical Chemistry C\",\"volume\":\"11 1\",\"pages\":\"\"},\"PeriodicalIF\":3.4000,\"publicationDate\":\"2024-10-29\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"The Journal of Physical Chemistry C\",\"FirstCategoryId\":\"1\",\"ListUrlMain\":\"https://doi.org/10.1021/acs.jpcc.4c04669\",\"RegionNum\":3,\"RegionCategory\":\"化学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q2\",\"JCRName\":\"CHEMISTRY, PHYSICAL\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"The Journal of Physical Chemistry C","FirstCategoryId":"1","ListUrlMain":"https://doi.org/10.1021/acs.jpcc.4c04669","RegionNum":3,"RegionCategory":"化学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q2","JCRName":"CHEMISTRY, PHYSICAL","Score":null,"Total":0}
引用次数: 0
摘要
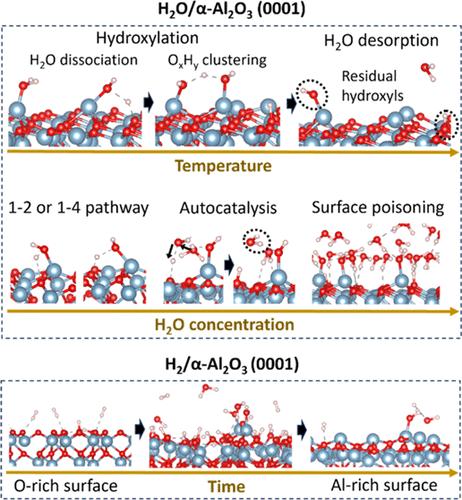
我们开发了 Al/O/H ReaxFF 力场,用于探索 H2O/H2 气相环境中 α-Al2O3 表面的化学反应。该力场可生成具有不同端点(Al 或 O-)的 A-、C-、R- 和 M-平面的表面能谱,并预测 Al 端点 α-Al2O3 (0001) 上水解的热力学和动力学行为,这与量子化学研究相一致。对 H2O/α-Al2O3 (0001) 的分子动力学(MD)模拟显示,水的自催化作用在加速铝端化 α-Al2O3 (0001) 上的 H2O 解离过程中发挥了重要作用。与 50% Al 端接的表面相比,100% Al 端接的表面在低至 350 K 的温度下更容易羟基化,更依赖于 OxHy 聚集机制而不是完全的 H2O 解离,一旦加热到 500 K 或更高温度,解吸的 H2O 分子明显增多。但在这两种情况下,加热都无法消除表面羟基,因此通过暴露于 H2O 而获得类似吉布斯特的表面是不太可能的。由于存在不规则的空位缺陷,以随机分布的表面铝物种为端点的 α-Al2O3 (0001) 上的 H2O 解离偏离了 1-2 和 1-4 路径,而且随机表面似乎比具有相同表面铝覆盖率的有序表面对 H2O 更敏感。对 H2/α-Al2O3 的模拟表明,密集的表面 O 覆盖和低热力学表面稳定性的结合会导致 H2 解离动力学的升高。为了加速 100% O 端接的 α-Al2O3 (0001) 在接触 H2 气体时的表面 O 清除,我们降低了 H-H σ 键的能量参数,相当于在模拟过程中将 H2 解离障碍降低了 ∼ 19.4 kcal/mol。1.5 ns 后,表面终结与 100% Al 终结相当,但保留了少量羟基。该力场揭示了 α-Al2O3 晶面和表面终止如何影响 H2O/H2 气体分子的解离行为,为今后针对蓝宝石薄膜外延的力场开发奠定了基础。
ReaxFF Study of Surface Chemical Reactions between α-Al2O3 Substrates and H2O/H2 Gas-Phase Molecules
We developed an Al/O/H ReaxFF force field to explore chemical reactions on α-Al2O3 surfaces in H2O/H2 gas-phase environments. This force field generates surface energy profiles of A-, C-, R-, and M-planes with various terminations (Al- or O-) and predicts the thermodynamic and kinetic behaviors of hydrolysis on Al-terminated α-Al2O3 (0001), consistent with quantum chemical studies. Molecular dynamics (MD) simulations of H2O/α-Al2O3 (0001) reveal that water autocatalysis plays a significant role in accelerating H2O dissociations on Al-terminated α-Al2O3 (0001). Compared with the 50% Al-terminated surface, the 100% Al-terminated surface becomes more easily hydroxylated at temperatures as low as 350 K, relying more on an OxHy clustering mechanism than complete H2O dissociations, and desorbs significantly more H2O molecules once heated up to 500 K or higher. But heating cannot eliminate surface hydroxyls for either case, and achieving a Gibbsite-like surface by H2O exposure is unlikely. H2O dissociations on α-Al2O3 (0001) terminated with randomly distributed surface Al species deviate from 1–2 and 1–4 pathways due to irregular vacancy defects, and a random surface appears to be more reactive to H2O than the ordered one with the same surface Al coverage. Simulations of H2/α-Al2O3 suggest that the combination of a dense surface O coverage and a low thermodynamic surface stability leads to elevated H2 dissociation kinetics. To accelerate the surface O removals of 100% O-terminated α-Al2O3 (0001) in H2 gas exposure, we reduced the H–H σ bond energy parameter, equivalent to lowering the H2 dissociation barrier by ∼ 19.4 kcal/mol during the simulation. After ∼ 1.5 ns, the surface termination became comparable to the 100% Al-terminated one but retained a small quantity of hydroxyls. This force field reveals how the α-Al2O3 crystallographic plane and the surface termination influence the dissociation behaviors of H2O/H2 gas molecules and lays the foundation for future force field developments targeted at thin film epitaxy on sapphire.
期刊介绍:
The Journal of Physical Chemistry A/B/C is devoted to reporting new and original experimental and theoretical basic research of interest to physical chemists, biophysical chemists, and chemical physicists.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
