{"title":"通过网络沙普利值分析发现乳腺癌抗 PD1 反应的可解释生物标志物","authors":"Chenxi Sun, Zhi-Ping Liu","doi":"10.1016/j.cmpb.2024.108481","DOIUrl":null,"url":null,"abstract":"<div><h3>Background and objective</h3><div>Immunotherapy holds promise in enhancing pathological complete response rates in breast cancer, albeit confined to a select cohort of patients. Consequently, pinpointing factors predictive of treatment responsiveness is of paramount importance. Gene expression and regulation, inherently operating within intricate networks, constitute fundamental molecular machinery for cellular processes and often serve as robust biomarkers. Nevertheless, contemporary feature selection approaches grapple with two key challenges: opacity in modeling and scarcity in accounting for gene-gene interactions</div></div><div><h3>Methods</h3><div>To address these limitations, we devise a novel feature selection methodology grounded in cooperative game theory, harmoniously integrating with sophisticated machine learning models. This approach identifies interconnected gene regulatory network biomarker modules with priori genetic linkage architecture. Specifically, we leverage Shapley values on network to quantify feature importance, while strategically constraining their integration based on network expansion principles and nodal adjacency, thereby fostering enhanced interpretability in feature selection. We apply our methods to a publicly available single-cell RNA sequencing dataset of breast cancer immunotherapy responses, using the identified feature gene set as biomarkers. Functional enrichment analysis with independent validations further illustrates their effective predictive performance</div></div><div><h3>Results</h3><div>We demonstrate the sophistication and excellence of the proposed method in data with network structure. It unveiled a cohesive biomarker module encompassing 27 genes for immunotherapy response. Notably, this module proves adept at precisely predicting anti-PD1 therapeutic outcomes in breast cancer patients with classification accuracy of 0.905 and AUC value of 0.971, underscoring its unique capacity to illuminate gene functionalities</div></div><div><h3>Conclusion</h3><div>The proposed method is effective for identifying network module biomarkers, and the detected anti-PD1 response biomarkers can enrich our understanding of the underlying physiological mechanisms of immunotherapy, which have a promising application for realizing precision medicine.</div></div>","PeriodicalId":10624,"journal":{"name":"Computer methods and programs in biomedicine","volume":"257 ","pages":"Article 108481"},"PeriodicalIF":4.8000,"publicationDate":"2024-12-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"","citationCount":"0","resultStr":"{\"title\":\"Discovering explainable biomarkers for breast cancer anti-PD1 response via network Shapley value analysis\",\"authors\":\"Chenxi Sun, Zhi-Ping Liu\",\"doi\":\"10.1016/j.cmpb.2024.108481\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<div><h3>Background and objective</h3><div>Immunotherapy holds promise in enhancing pathological complete response rates in breast cancer, albeit confined to a select cohort of patients. Consequently, pinpointing factors predictive of treatment responsiveness is of paramount importance. Gene expression and regulation, inherently operating within intricate networks, constitute fundamental molecular machinery for cellular processes and often serve as robust biomarkers. Nevertheless, contemporary feature selection approaches grapple with two key challenges: opacity in modeling and scarcity in accounting for gene-gene interactions</div></div><div><h3>Methods</h3><div>To address these limitations, we devise a novel feature selection methodology grounded in cooperative game theory, harmoniously integrating with sophisticated machine learning models. This approach identifies interconnected gene regulatory network biomarker modules with priori genetic linkage architecture. Specifically, we leverage Shapley values on network to quantify feature importance, while strategically constraining their integration based on network expansion principles and nodal adjacency, thereby fostering enhanced interpretability in feature selection. We apply our methods to a publicly available single-cell RNA sequencing dataset of breast cancer immunotherapy responses, using the identified feature gene set as biomarkers. Functional enrichment analysis with independent validations further illustrates their effective predictive performance</div></div><div><h3>Results</h3><div>We demonstrate the sophistication and excellence of the proposed method in data with network structure. It unveiled a cohesive biomarker module encompassing 27 genes for immunotherapy response. Notably, this module proves adept at precisely predicting anti-PD1 therapeutic outcomes in breast cancer patients with classification accuracy of 0.905 and AUC value of 0.971, underscoring its unique capacity to illuminate gene functionalities</div></div><div><h3>Conclusion</h3><div>The proposed method is effective for identifying network module biomarkers, and the detected anti-PD1 response biomarkers can enrich our understanding of the underlying physiological mechanisms of immunotherapy, which have a promising application for realizing precision medicine.</div></div>\",\"PeriodicalId\":10624,\"journal\":{\"name\":\"Computer methods and programs in biomedicine\",\"volume\":\"257 \",\"pages\":\"Article 108481\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-12-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Computer methods and programs in biomedicine\",\"FirstCategoryId\":\"5\",\"ListUrlMain\":\"https://www.sciencedirect.com/science/article/pii/S0169260724004747\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"2024/10/26 0:00:00\",\"PubModel\":\"Epub\",\"JCR\":\"Q1\",\"JCRName\":\"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Computer methods and programs in biomedicine","FirstCategoryId":"5","ListUrlMain":"https://www.sciencedirect.com/science/article/pii/S0169260724004747","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"2024/10/26 0:00:00","PubModel":"Epub","JCR":"Q1","JCRName":"COMPUTER SCIENCE, INTERDISCIPLINARY APPLICATIONS","Score":null,"Total":0}
Discovering explainable biomarkers for breast cancer anti-PD1 response via network Shapley value analysis
Background and objective
Immunotherapy holds promise in enhancing pathological complete response rates in breast cancer, albeit confined to a select cohort of patients. Consequently, pinpointing factors predictive of treatment responsiveness is of paramount importance. Gene expression and regulation, inherently operating within intricate networks, constitute fundamental molecular machinery for cellular processes and often serve as robust biomarkers. Nevertheless, contemporary feature selection approaches grapple with two key challenges: opacity in modeling and scarcity in accounting for gene-gene interactions
Methods
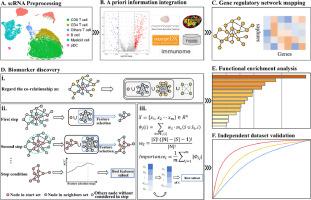
To address these limitations, we devise a novel feature selection methodology grounded in cooperative game theory, harmoniously integrating with sophisticated machine learning models. This approach identifies interconnected gene regulatory network biomarker modules with priori genetic linkage architecture. Specifically, we leverage Shapley values on network to quantify feature importance, while strategically constraining their integration based on network expansion principles and nodal adjacency, thereby fostering enhanced interpretability in feature selection. We apply our methods to a publicly available single-cell RNA sequencing dataset of breast cancer immunotherapy responses, using the identified feature gene set as biomarkers. Functional enrichment analysis with independent validations further illustrates their effective predictive performance
Results
We demonstrate the sophistication and excellence of the proposed method in data with network structure. It unveiled a cohesive biomarker module encompassing 27 genes for immunotherapy response. Notably, this module proves adept at precisely predicting anti-PD1 therapeutic outcomes in breast cancer patients with classification accuracy of 0.905 and AUC value of 0.971, underscoring its unique capacity to illuminate gene functionalities
Conclusion
The proposed method is effective for identifying network module biomarkers, and the detected anti-PD1 response biomarkers can enrich our understanding of the underlying physiological mechanisms of immunotherapy, which have a promising application for realizing precision medicine.
期刊介绍:
To encourage the development of formal computing methods, and their application in biomedical research and medical practice, by illustration of fundamental principles in biomedical informatics research; to stimulate basic research into application software design; to report the state of research of biomedical information processing projects; to report new computer methodologies applied in biomedical areas; the eventual distribution of demonstrable software to avoid duplication of effort; to provide a forum for discussion and improvement of existing software; to optimize contact between national organizations and regional user groups by promoting an international exchange of information on formal methods, standards and software in biomedicine.
Computer Methods and Programs in Biomedicine covers computing methodology and software systems derived from computing science for implementation in all aspects of biomedical research and medical practice. It is designed to serve: biochemists; biologists; geneticists; immunologists; neuroscientists; pharmacologists; toxicologists; clinicians; epidemiologists; psychiatrists; psychologists; cardiologists; chemists; (radio)physicists; computer scientists; programmers and systems analysts; biomedical, clinical, electrical and other engineers; teachers of medical informatics and users of educational software.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
