{"title":"通过开发疾病特异性生长曲线图解读韩国索托斯综合征患儿的生长模式","authors":"Naye Choi, Hwa Young Kim, Jung Min Ko","doi":"10.1002/mgg3.70028","DOIUrl":null,"url":null,"abstract":"<p><strong>Background: </strong>Sotos syndrome (SS) is a rare disorder characterized by overgrowth, distinctive facial features, and intellectual disability that is primarily caused by NSD1 pathogenic variants or 5q35 microdeletions.</p><p><strong>Methods: </strong>We retrospectively analyzed the clinical characteristics and 339 anthropometric measurements over an average of 4.3 years of follow-up in 57 Korean children with SS. Sex-specific percentile curves for height, weight, and head circumference were developed using a generalized additive model that included factors such as location, scale, and shape.</p><p><strong>Results: </strong>Males with SS demonstrated higher height before the age of 12.0, greater weight before 10.0, and larger head circumference before 15.5 compared to age- and sex-matched controls. Females with SS displayed higher height before 17.0, greater weight before 10.5, and larger head circumference before 12.0 compared to controls. Bone age was advanced compared to chronological age in 40% of males and 8% of females at their last visit. The predicted and target adult heights were not significantly different between groups. In subgroup analysis, the intragenic variant group (n = 48) showed a higher mean standard deviation score of height and weight in males, and head circumference in females compared to the microdeletion group (n = 9).</p><p><strong>Conclusions: </strong>Korean children with genetically confirmed SS exhibited overgrowth in height, weight, and head circumference. Overgrowth phenotypes were more prominent in patients with NSD1 intragenic variants than in those with microdeletions. This is the first study to provide reference data on the growth of Korean children with SS.</p>","PeriodicalId":18852,"journal":{"name":"Molecular Genetics & Genomic Medicine","volume":"12 11","pages":"e70028"},"PeriodicalIF":1.6000,"publicationDate":"2024-11-01","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11532833/pdf/","citationCount":"0","resultStr":"{\"title\":\"Deciphering Growth Patterns in Korean Children With Sotos Syndrome Through the Development of a Disease-Specific Growth Chart.\",\"authors\":\"Naye Choi, Hwa Young Kim, Jung Min Ko\",\"doi\":\"10.1002/mgg3.70028\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><strong>Background: </strong>Sotos syndrome (SS) is a rare disorder characterized by overgrowth, distinctive facial features, and intellectual disability that is primarily caused by NSD1 pathogenic variants or 5q35 microdeletions.</p><p><strong>Methods: </strong>We retrospectively analyzed the clinical characteristics and 339 anthropometric measurements over an average of 4.3 years of follow-up in 57 Korean children with SS. Sex-specific percentile curves for height, weight, and head circumference were developed using a generalized additive model that included factors such as location, scale, and shape.</p><p><strong>Results: </strong>Males with SS demonstrated higher height before the age of 12.0, greater weight before 10.0, and larger head circumference before 15.5 compared to age- and sex-matched controls. Females with SS displayed higher height before 17.0, greater weight before 10.5, and larger head circumference before 12.0 compared to controls. Bone age was advanced compared to chronological age in 40% of males and 8% of females at their last visit. The predicted and target adult heights were not significantly different between groups. In subgroup analysis, the intragenic variant group (n = 48) showed a higher mean standard deviation score of height and weight in males, and head circumference in females compared to the microdeletion group (n = 9).</p><p><strong>Conclusions: </strong>Korean children with genetically confirmed SS exhibited overgrowth in height, weight, and head circumference. Overgrowth phenotypes were more prominent in patients with NSD1 intragenic variants than in those with microdeletions. This is the first study to provide reference data on the growth of Korean children with SS.</p>\",\"PeriodicalId\":18852,\"journal\":{\"name\":\"Molecular Genetics & Genomic Medicine\",\"volume\":\"12 11\",\"pages\":\"e70028\"},\"PeriodicalIF\":1.6000,\"publicationDate\":\"2024-11-01\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11532833/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"Molecular Genetics & Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1002/mgg3.70028\",\"RegionNum\":4,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q4\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"Molecular Genetics & Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1002/mgg3.70028","RegionNum":4,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q4","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
引用次数: 0
摘要
背景:索托斯综合征(SS)是一种罕见的疾病,主要由 NSD1 致病变体或 5q35 微缺失引起,以发育过度、独特的面部特征和智力障碍为特征:索托斯综合征(SS)是一种罕见的疾病,主要由NSD1致病变体或5q35微缺失引起,以发育过度、独特的面部特征和智力障碍为特征:我们对 57 名韩国 SS 儿童平均 4.3 年的临床特征和 339 项人体测量数据进行了回顾性分析。结果:患有 SS 的男性在身高、体重和头围方面表现得更高,而患有 SS 的女性在身高、体重和头围方面表现得更低:结果:与年龄和性别匹配的对照组相比,患有 SS 的男性在 12.0 岁前身高更高,在 10.0 岁前体重更大,在 15.5 岁前头围更大。与对照组相比,患有 SS 的女性在 17.0 岁前身高更高、体重在 10.5 岁前更重、头围在 12.0 岁前更大。在最后一次就诊时,40%的男性和 8%的女性的骨龄比实际年龄提前。各组之间的预测身高和目标成人身高没有显著差异。在亚组分析中,与微缺失组(n = 9)相比,基因内变异组(n = 48)的男性身高和体重以及女性头围的平均标准偏差得分更高:结论:经遗传学证实,患有 SS 的韩国儿童在身高、体重和头围方面表现出过度生长。与微缺失组相比,NSD1 基因内变异患者的过度生长表型更为突出。这是第一项提供韩国 SS 儿童生长参考数据的研究。
Deciphering Growth Patterns in Korean Children With Sotos Syndrome Through the Development of a Disease-Specific Growth Chart.
Background: Sotos syndrome (SS) is a rare disorder characterized by overgrowth, distinctive facial features, and intellectual disability that is primarily caused by NSD1 pathogenic variants or 5q35 microdeletions.
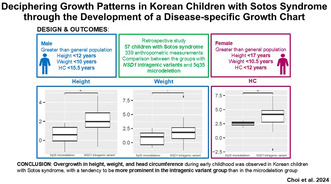
Methods: We retrospectively analyzed the clinical characteristics and 339 anthropometric measurements over an average of 4.3 years of follow-up in 57 Korean children with SS. Sex-specific percentile curves for height, weight, and head circumference were developed using a generalized additive model that included factors such as location, scale, and shape.
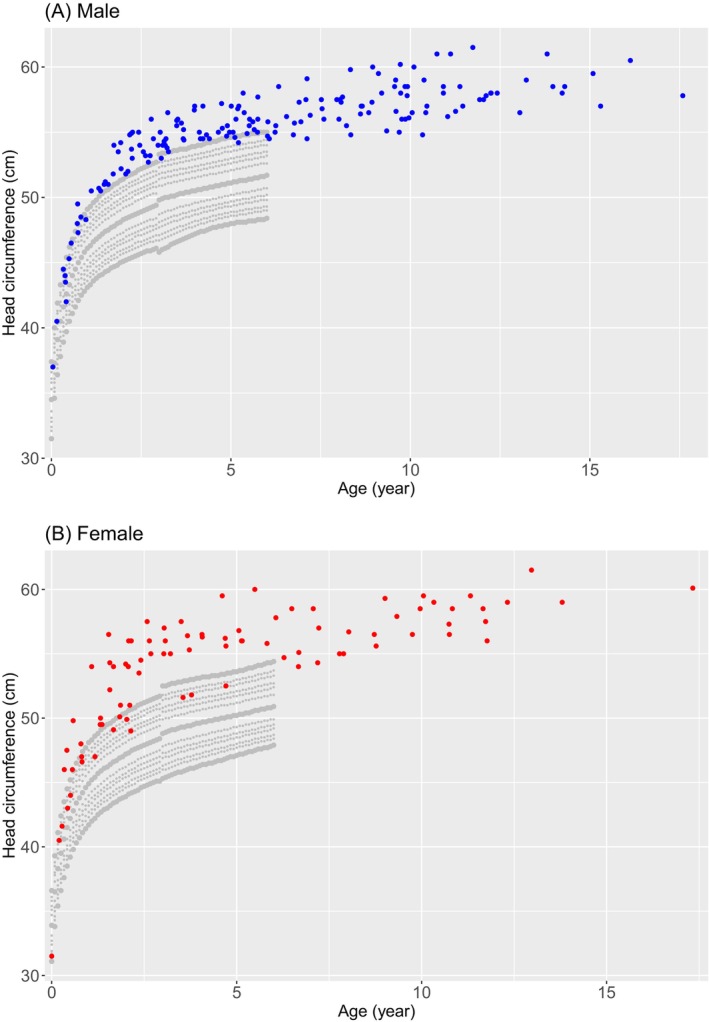
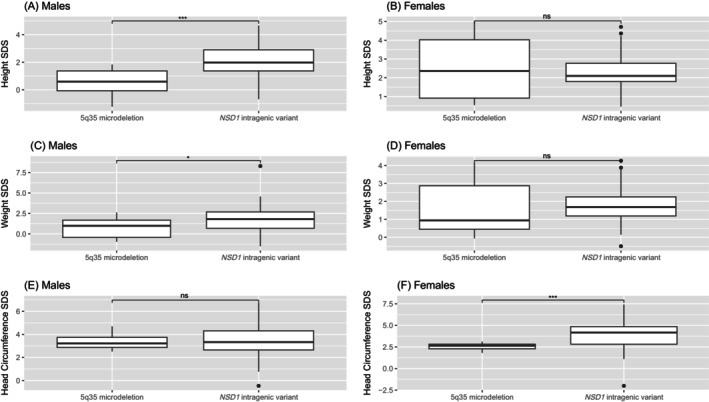
Results: Males with SS demonstrated higher height before the age of 12.0, greater weight before 10.0, and larger head circumference before 15.5 compared to age- and sex-matched controls. Females with SS displayed higher height before 17.0, greater weight before 10.5, and larger head circumference before 12.0 compared to controls. Bone age was advanced compared to chronological age in 40% of males and 8% of females at their last visit. The predicted and target adult heights were not significantly different between groups. In subgroup analysis, the intragenic variant group (n = 48) showed a higher mean standard deviation score of height and weight in males, and head circumference in females compared to the microdeletion group (n = 9).
Conclusions: Korean children with genetically confirmed SS exhibited overgrowth in height, weight, and head circumference. Overgrowth phenotypes were more prominent in patients with NSD1 intragenic variants than in those with microdeletions. This is the first study to provide reference data on the growth of Korean children with SS.
期刊介绍:
Molecular Genetics & Genomic Medicine is a peer-reviewed journal for rapid dissemination of quality research related to the dynamically developing areas of human, molecular and medical genetics. The journal publishes original research articles covering findings in phenotypic, molecular, biological, and genomic aspects of genomic variation, inherited disorders and birth defects. The broad publishing spectrum of Molecular Genetics & Genomic Medicine includes rare and common disorders from diagnosis to treatment. Examples of appropriate articles include reports of novel disease genes, functional studies of genetic variants, in-depth genotype-phenotype studies, genomic analysis of inherited disorders, molecular diagnostic methods, medical bioinformatics, ethical, legal, and social implications (ELSI), and approaches to clinical diagnosis. Molecular Genetics & Genomic Medicine provides a scientific home for next generation sequencing studies of rare and common disorders, which will make research in this fascinating area easily and rapidly accessible to the scientific community. This will serve as the basis for translating next generation sequencing studies into individualized diagnostics and therapeutics, for day-to-day medical care.
Molecular Genetics & Genomic Medicine publishes original research articles, reviews, and research methods papers, along with invited editorials and commentaries. Original research papers must report well-conducted research with conclusions supported by the data presented.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
