Simo Li, Sanami Takada, Ghada M H Abdel-Salam, Mohamed S Abdel-Hamid, Maha S Zaki, Mahmoud Y Issa, Aida M S Salem, Eriko Koshimizu, Atsushi Fujita, Ryoko Fukai, Toshio Ohshima, Naomichi Matsumoto, Noriko Miyake
{"title":"GON4L的双倍功能缺失变体会导致小头畸形和脑结构异常。","authors":"Simo Li, Sanami Takada, Ghada M H Abdel-Salam, Mohamed S Abdel-Hamid, Maha S Zaki, Mahmoud Y Issa, Aida M S Salem, Eriko Koshimizu, Atsushi Fujita, Ryoko Fukai, Toshio Ohshima, Naomichi Matsumoto, Noriko Miyake","doi":"10.1038/s41525-024-00437-5","DOIUrl":null,"url":null,"abstract":"<p><p>We identified two homozygous truncating variants in GON4L [NM_001282860.2:c.62_63del, p.(Gln21Argfs*12) and c.5517+1G>A] in two unrelated families who presented prenatal-onset growth impairment, microcephaly, characteristic face, situs inversus, and developmental delay. The frameshift variant is predicted to invoke nonsense-mediated mRNA decay of all five known GON4L isoforms resulting in the complete loss of GON4L function. The splice site variant located at a region specific to the longer isoforms; therefore, defects of long GON4L isoforms may explain the phenotypes observed in the three patients. Knockdown of Gon4l in rat PC12 cells suppressed neurite outgrowth in vitro. gon4lb knockdown and knockout zebrafish successfully recapitulated the patients' phenotypes including craniofacial abnormalities. We also observed situs inversus in gon4lb-knockout zebrafish embryo. To our knowledge, the relationship between craniofacial abnormalities or situs inversus and gon4lb has not been reported before. Thus, our data provide evidence that GON4L is involved in craniofacial and left-right patterning during development.</p>","PeriodicalId":19273,"journal":{"name":"NPJ Genomic Medicine","volume":"9 1","pages":"55"},"PeriodicalIF":4.8000,"publicationDate":"2024-11-05","publicationTypes":"Journal Article","fieldsOfStudy":null,"isOpenAccess":false,"openAccessPdf":"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11538285/pdf/","citationCount":"0","resultStr":"{\"title\":\"Biallelic loss-of-function variants in GON4L cause microcephaly and brain structure abnormalities.\",\"authors\":\"Simo Li, Sanami Takada, Ghada M H Abdel-Salam, Mohamed S Abdel-Hamid, Maha S Zaki, Mahmoud Y Issa, Aida M S Salem, Eriko Koshimizu, Atsushi Fujita, Ryoko Fukai, Toshio Ohshima, Naomichi Matsumoto, Noriko Miyake\",\"doi\":\"10.1038/s41525-024-00437-5\",\"DOIUrl\":null,\"url\":null,\"abstract\":\"<p><p>We identified two homozygous truncating variants in GON4L [NM_001282860.2:c.62_63del, p.(Gln21Argfs*12) and c.5517+1G>A] in two unrelated families who presented prenatal-onset growth impairment, microcephaly, characteristic face, situs inversus, and developmental delay. The frameshift variant is predicted to invoke nonsense-mediated mRNA decay of all five known GON4L isoforms resulting in the complete loss of GON4L function. The splice site variant located at a region specific to the longer isoforms; therefore, defects of long GON4L isoforms may explain the phenotypes observed in the three patients. Knockdown of Gon4l in rat PC12 cells suppressed neurite outgrowth in vitro. gon4lb knockdown and knockout zebrafish successfully recapitulated the patients' phenotypes including craniofacial abnormalities. We also observed situs inversus in gon4lb-knockout zebrafish embryo. To our knowledge, the relationship between craniofacial abnormalities or situs inversus and gon4lb has not been reported before. Thus, our data provide evidence that GON4L is involved in craniofacial and left-right patterning during development.</p>\",\"PeriodicalId\":19273,\"journal\":{\"name\":\"NPJ Genomic Medicine\",\"volume\":\"9 1\",\"pages\":\"55\"},\"PeriodicalIF\":4.8000,\"publicationDate\":\"2024-11-05\",\"publicationTypes\":\"Journal Article\",\"fieldsOfStudy\":null,\"isOpenAccess\":false,\"openAccessPdf\":\"https://www.ncbi.nlm.nih.gov/pmc/articles/PMC11538285/pdf/\",\"citationCount\":\"0\",\"resultStr\":null,\"platform\":\"Semanticscholar\",\"paperid\":null,\"PeriodicalName\":\"NPJ Genomic Medicine\",\"FirstCategoryId\":\"3\",\"ListUrlMain\":\"https://doi.org/10.1038/s41525-024-00437-5\",\"RegionNum\":2,\"RegionCategory\":\"医学\",\"ArticlePicture\":[],\"TitleCN\":null,\"AbstractTextCN\":null,\"PMCID\":null,\"EPubDate\":\"\",\"PubModel\":\"\",\"JCR\":\"Q1\",\"JCRName\":\"GENETICS & HEREDITY\",\"Score\":null,\"Total\":0}","platform":"Semanticscholar","paperid":null,"PeriodicalName":"NPJ Genomic Medicine","FirstCategoryId":"3","ListUrlMain":"https://doi.org/10.1038/s41525-024-00437-5","RegionNum":2,"RegionCategory":"医学","ArticlePicture":[],"TitleCN":null,"AbstractTextCN":null,"PMCID":null,"EPubDate":"","PubModel":"","JCR":"Q1","JCRName":"GENETICS & HEREDITY","Score":null,"Total":0}
Biallelic loss-of-function variants in GON4L cause microcephaly and brain structure abnormalities.
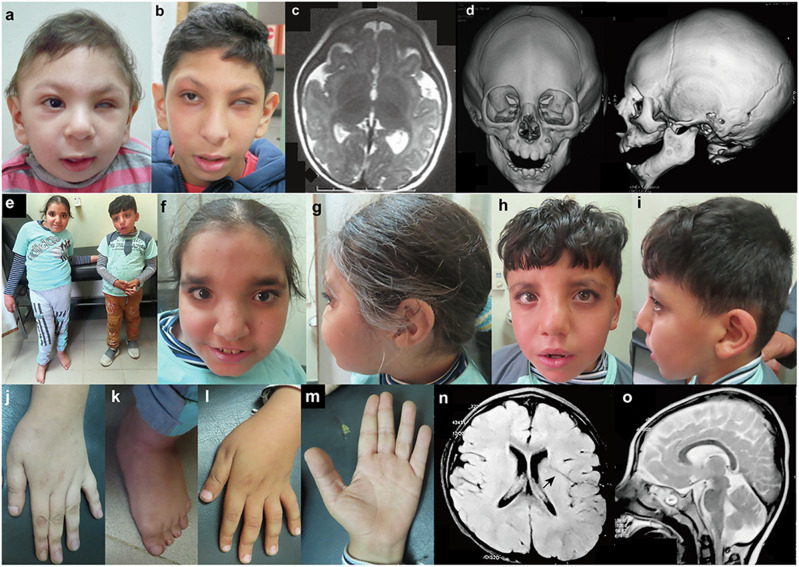
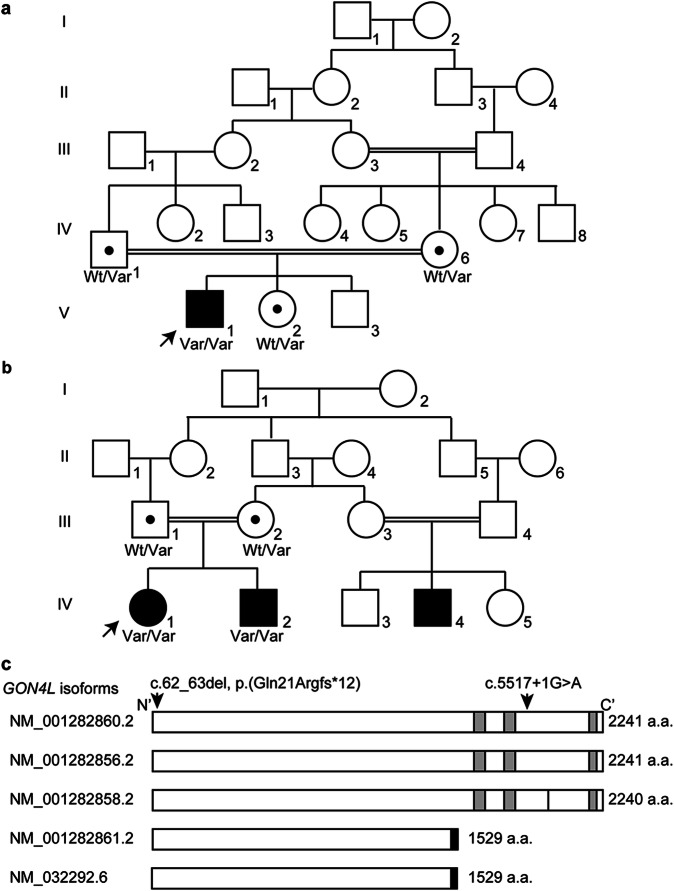
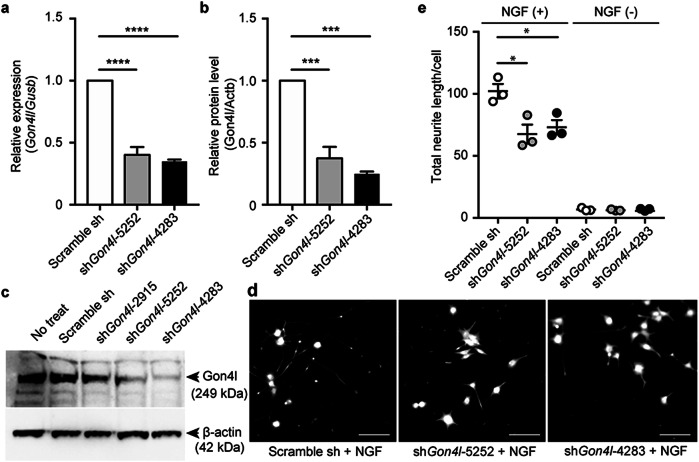
We identified two homozygous truncating variants in GON4L [NM_001282860.2:c.62_63del, p.(Gln21Argfs*12) and c.5517+1G>A] in two unrelated families who presented prenatal-onset growth impairment, microcephaly, characteristic face, situs inversus, and developmental delay. The frameshift variant is predicted to invoke nonsense-mediated mRNA decay of all five known GON4L isoforms resulting in the complete loss of GON4L function. The splice site variant located at a region specific to the longer isoforms; therefore, defects of long GON4L isoforms may explain the phenotypes observed in the three patients. Knockdown of Gon4l in rat PC12 cells suppressed neurite outgrowth in vitro. gon4lb knockdown and knockout zebrafish successfully recapitulated the patients' phenotypes including craniofacial abnormalities. We also observed situs inversus in gon4lb-knockout zebrafish embryo. To our knowledge, the relationship between craniofacial abnormalities or situs inversus and gon4lb has not been reported before. Thus, our data provide evidence that GON4L is involved in craniofacial and left-right patterning during development.
NPJ Genomic MedicineBiochemistry, Genetics and Molecular Biology-Molecular Biology
CiteScore
9.40
自引率
1.90%
发文量
67
审稿时长
17 weeks
期刊介绍:
npj Genomic Medicine is an international, peer-reviewed journal dedicated to publishing the most important scientific advances in all aspects of genomics and its application in the practice of medicine.
The journal defines genomic medicine as "diagnosis, prognosis, prevention and/or treatment of disease and disorders of the mind and body, using approaches informed or enabled by knowledge of the genome and the molecules it encodes." Relevant and high-impact papers that encompass studies of individuals, families, or populations are considered for publication. An emphasis will include coupling detailed phenotype and genome sequencing information, both enabled by new technologies and informatics, to delineate the underlying aetiology of disease. Clinical recommendations and/or guidelines of how that data should be used in the clinical management of those patients in the study, and others, are also encouraged.




 分享
分享
 求助内容:
求助内容: 应助结果提醒方式:
应助结果提醒方式: 扫码关注我们
扫码关注我们
